|
To view this email as a web page, click here. |
 |
|
Welcome
Specifying the right set of modifications is a critical part of obtaining high quality search results, and we have a few suggestions to help.
This month's highlighted publication shows an approach for characterizing and quantifying difficult-to-analyze proteins from the SNARE complex.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please send us a PDF or a URL.
If you are running Mascot Server 2.5, you'll be interested to know that a patch has just been released.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
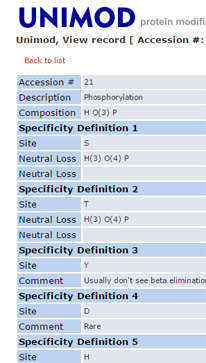
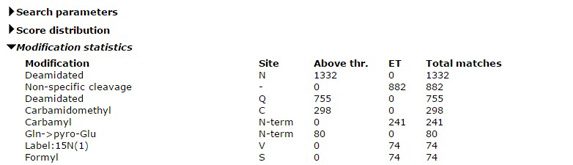
Why can't I select more than 9 variable modifications?
It is always tempting to select all the modifications that might be present in the sample as variable modifications for a search, but it is a temptation that should be resisted. The default limit on the number of variable modifications is 9. This limit can be increased by making a change in the global configuration file, but doing so is usually a mistake.
Variable modifications are most useful for relatively abundant modifications, that occur on at least 10% of peptides. During a search, Mascot looks for all the combinations of the variable modifications that fit to the peptide mass, then looks at all the arrangements of these to see which gives the best fit to the MS/MS fragments. Each additional variable modification leads to a large increase in the size of the search space, especially for large peptides. The larger the search space, the longer the search takes and the higher the score required to get a significant match. If any of the variable modifications are relatively rare, you end up waiting longer to get fewer matches.
All post-translational modifications are rare at the proteome level. If you are analysing a whole cell lysate, the smart way to find post-translational modifications is with an error tolerant search, which tests all known modifications, serially. It only misses peptides that carry two or more different, unsuspected modifications, which has negligible impact on overall sensitivity. It isn't a universal solution, and cannot be used with isolated peptides, such as MHC peptides, or where the sample has been enriched in a particular class of peptide or protein, such phosphopeptides or glycopeptides. But, in most cases, it is the best place to start, if only to see what modifications should be considered in a more targeted experiment.
More information about modifications in Mascot searches can be found in two recent blog articles - Modifications round-up, part 1 and part 2. |
 |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
Targeting Synaptic Pathology with a Novel Affinity Mass Spectrometry Approach
Ann Brinkmalm, Gunnar Brinkmalm, William G. Honer, Julie A. Moreno, Joel Jakobsson,
Giovanna R. Mallucci, Henrik Zetterberg, Kaj Blennow, and Annika Öhrfelt.
Molecular & Cellular Proteomics October 1, 2014, Vol. 13, 2584-2592
This study presents a method for characterizing and quantifying proteins in the SNARE complex, which are involved in presynaptic regulation and may play a role in early stage dysfunction in Alzheimer's disease. These proteins are difficult to analyze by mass spectrometry as they are membrane-associated and strongly form complexes.
Combining affinity purification by immunoprecipitation and mass spectrometry, the authors showed reduced levels of the SNARE proteins in brain tissue from AD patients as well as in prion-infected mice. The particular advantage of the MS-based method over immunological assays is that all SNARE proteins, including modified forms, can be quantified simultaneously.
|
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by AB Sciex, Agilent, Bruker, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|

