|
To view this email as a web page, click here. |
 |
|
Welcome
Is a feature driven approach to discovery quantitation more efficient than identifying everything first? Probably not, unless you are using 2D gels.
This month's highlighted publication shows a novel way of unravelling some of the detailed interactions within the proteasome.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
There is a reminder about our symposium at the upcoming Proteomic Forum 2015 in Berlin on March 22-25 and a new section: Mascot Tip of the Month.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
Quantify then identify or identify then quantify?
Many studies look at variation in protein expression as a function of disease or stress.
In such cases, why waste time acquiring MS/MS to identify peptides from proteins that are not changing?
Wouldn't it be more efficient to focus attention on the regulated proteins?
Yes, if you are using a 2D gel to visualise the proteins, so that those varying in abundance can be
excised, digested, and analyzed. Unfortunately, such an approach doesn't work in the context of shotgun proteomics.
It might be perfectly feasible to select peptides that vary in abundance for MS/MS,
but one peptide says little or nothing about the abundance of the protein.
To read a detailed discussion of this topic, click here. |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
Deciphering Preferential
Interactions within Supramolecular Protein Complexes: the Proteasome Case
Bertrand Fabre, Thomas Lambour, Luc Garrigues, François Amalric, Nathalie Vigneron,
Thomas Menneteau, Alexandre Stella, Bernard Monsarrat, Benoît Van den Eynde, Odile Burlet-Schiltz,
& Marie-Pierre Bousquet-Dubouch
Molecular Systems Biology (2015) 11: 771
Proteasomes are dynamic structures and adapt to their diverse biological contexts by
changing their overall subunit composition and association with regulatory particles and interacting proteins.
This high level of proteasome diversity could reflect specialized functions of each individual proteasome form.
This paper demonstrates an approach to decipher proteasome heterogeneity through label-free
quantitative proteomics. The authors analyzed sub-complex composition by combining affinity purification and
protein abundance correlation profiling with high-resolution mass spectrometry. By correlating proteins
abundances across a large set of 24 proteasome samples immunopurified from nine different human cell lines,
they observed that the two main 20S proteasome subtypes, sP20S and iP20S, interact with a different
subset of regulators. This approach could be applied to other important large complexes.
|
 |
|
|
|
|
|
|
|
Large Scale Proteomics symposium at Proteomic Forum 2015
Don't miss our lunch-time symposium - Large Scale Proteomics - during the upcoming
Proteomic Forum 2015,
March 22-25 in Berlin, Germany. The event features two presentations and is open to registered attendees.
- Analysis of the human proteome in proteomicsDB, Bernhard Kuster, Technische Universitaet Muenchen
- Reporting complex quantitation data sets using Mascot Insight, Patrick Emery, Matrix Science Ltd.
Places are limited, so early registration is advisable. |
 |
|
|
|
|
|
|
|
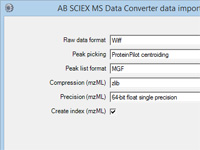
Mascot tip of the month
Did you realize you can configure almost any Windows executable that creates a peak list from a
raw file as a data import filter for Mascot Daemon? For more information, press F1 in Daemon and read the topic
In depth; Data import filters; Adding a new filter.
If you have difficulty with the configuration or if you think other Mascot Daemon users might want
to use the same utility, please send us details and we'll consider whether to add it to the standard configuration. |
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by AB Sciex, Agilent, Bruker, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
