|
To view this email as a web page, click here. |
 |
|
Welcome
We look at a new, open-source utility for deconvoluting DIA data to create a DDA-style peak list for searching with Mascot.
This month's highlighted publication discusses the process by which MHC Class I peptides are selected and presented to the cell surface.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month describes how to control the way in which Mascot Server re-centroids a peak list.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
DIA-Umpire enables database searching of DIA data
An increasing number of researchers are using data-independent acquisition (DIA) for targeted quantitation. DIA-Umpire is a new, open-source Java utility from the University of Michigan, described in
Nature Methods 12, 258-264 (2015), that deconvolutes DIA data to create a conventional, DDA-style peak list. This enables untargeted identification and quantitation without any reliance on spectral libraries. The software is intended to work with DIA from any instrument.
We tried processing and searching a file of Swath data, acquired on an AB Sciex 5600, and the results looked good. If you want to process and search a large number of files, it is possible, though not easy, to use the utility as a data import filter in Mascot Daemon 2.5.
More details in this recent blog article. |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
The first step of peptide selection in antigen presentation by MHC class I molecules
Malgorzata A. Garstka, Alexander Fish, Patrick H. N. Celie, Robbie P. Joosten, George M. C. Janssen, Ilana Berlin, Rieuwert Hoppes, Magda Stadnik, Lennert Janssen, Huib Ovaa, Peter A. van Veelen, Anastassis Perrakis, and Jacques Neefjes
PNAS (2015) 112: 1505-10
The major histocompatibility complex (MHC) class I molecules display a wide array of peptides to the cytotoxic T cells, providing for the immune system to attack the pathogens and mutated proteins. However there is a limited set of peptides that is presented, and how this selection occurs is the focus of this paper, in which the peptide on- and off-rates from the mouse MHC class I molecule H-2K were studied.
The authors found that these molecules initially bind a large number of peptides, but that this initial step is followed by a selection where the majority of these peptides are released. They found a strong temperature dependency by the H-2Kb peptidome from cells cultured at 26 °C versus 37 °C, suggesting that the MHC class I undergoes rounds of consideration and rejection until only high-affinity peptides are acquired. This mechanism simultaneously allows for a wide range of sampling while ensuring that only the best available options are chosen for presentation.
|
 |
|
|
|
|
|
|
|
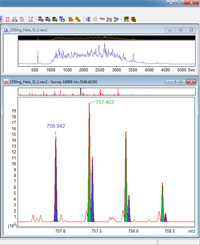
Mascot tip of the month
Mascot has always centroided the peak list to try to give reasonable results when profile data are uploaded. This has no effect on isolated peaks in a true peak list, but if there are two closely-spaced peaks within the centroiding window, they will be replaced by a single peak. As instruments capable of high resolution, high accuracy MS/MS data started to become more widely available, we recognized that this could be a problem, and made centroiding dependent on there being more than 1,000 peaks in the spectrum, which disables it for any true peak list. This change was introduced in Mascot 2.4.
If you have high resolution, high accuracy MS/MS data and are using Mascot 2.3 or earlier, re-centroiding may be a problem, particularly when it causes small shifts in the positions of reporter ion peaks (iTRAQ, TMT). You can disable centroiding by setting CentroidWidth in the Options section of mascot.dat to a very small value, e.g. 0.0001. If you do this, be aware that anyone in the habit of uploading profile data will suddenly find their search results become a whole lot worse! |
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by AB Sciex, Agilent, Bruker, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
