|
To view this email as a web page, click here. |
 |
|
Welcome
Is searching with a very wide mass tolerance a better way to find unsuspected modifications? Only in certain special cases, we suggest.
This month’s highlighted publication shows a new method to enrich for and quantify protein S-glutathionylation, which is important in helping cells tolerate stress.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month concerns searching data that mixes multiple ionization methods, such as alternate CID / ETD acquisition.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
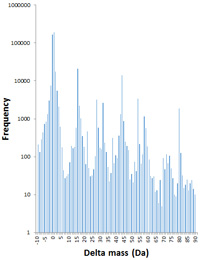
Mass-tolerant vs error tolerant search strategies
The Gygi lab. has proposed using a 500 Da mass tolerance as a way of matching peptides carrying unsuspected modifications. To understand the utility of this approach, we compared their results, published in Nature Biotechnology, with the more conventional error tolerant search.
For this comparison, we downloaded the original HEK 293 data set and used Mascot Daemon to automate peak picking of the files using Mascot Distiller, merged them, and submitted an error tolerant search to Mascot Server. As you might expect, many of the most abundant modifications, with more than 1000 instances each, appear in both sets of results, and are the "usual suspects".
Two of the most abundant delta masses reported by the mass-tolerant search cannot be explained, and it seems clear that more time and effort is required for results interpretation compared with an error tolerant search. In fact, the only modification assigned a structure or composition in the paper that is missing from Unimod is a series of polyalanine insertions found in ribosomal protein L14.
Possibly, the main application of the mass-tolerant search is characterising modifications on endogenous peptides, because an error tolerant search cannot be used for isolated peptides. Go here to read about the comparison in greater detail. |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
Proteome-wide identification and quantification of S-glutathionylation targets in mouse liver
David J. McGarry, Wenzhang Chen, Probir Chakravarty, Douglas L. Lamont, C. Roland Wolf, Colin J. Henderson
Biochemical Journal Jun 19, 2015, 469, 25-32
Protein S-glutathionylation is an important post-translational modification that maintains redox homeostasis in response to stress, but its physiological role is not well understood. In this study, the authors developed a robust and optimized methodology for high-accuracy quantification and identification of S-glutathionylated targets in an animal model. They also established a number of S-glutathionylation pathways related to energy metabolism.
Grx1 is used to derivatize free thiol groups from S-glutathionylated proteins, which are then isolated by binding to thiopropyl Sepharose 6B beads. Then they identified and quantified these in vivo targets of S-glutathionylation using isobaric tandem mass tags (TMTs) with six-fold multiplexing. After TMT labelling and nLC–MS/MS analysis, a total of 743 proteins were identified from mouse liver lysates. |
 |
|
|
|
|
|
|
|
Mascot tip of the month
The Instrument selection in a Mascot search determines which ion series are considered when matching MS/MS data. Looking at all possible
ion series would be an unnecessary increase in the size of the search space, leading to a reduction in discrimination.
The Instrument can be specified at search level or scan level, so you might think that there would be no difficulty searching an acquisition that combines different types of ionization, such as alternating CID and ETD. Unfortunately, none of the current generation of peak picking utilities are capable of entering this information into the peak list.
The work-around is to create an Instrument definition that is a super-set of the two. For CID (or HCD) and ETD, this means mainly b, c, y, and z ions. If your Mascot Server does not have a CID+ETD Instrument setting, you can easily add it using the configuration editor.
When performing peak picking, make sure you disable summing of scans based on precursor m/z and retention times. You'll get better results by keeping the two scan types separate. When the search is submitted, just select the combined instrument type from the drop down list of instruments. Peptides will be identified with predominately CID ion series or predominately ETD ion series. |
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
