|
To view this email as a web page, click here. |
 |
|
Welcome
Database search is mainly used for protein identification, but it can also assist with characterization of recombinant proteins.
In this month's featured publication, the authors elucidate histone modifications and truncations.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
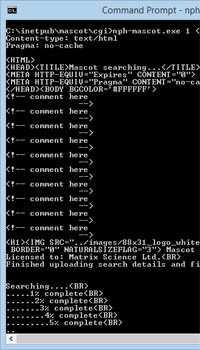
Mascot tip of the month describes how to submit a search at the command line when the peak list is too big for the web server to handle.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
Assuring protein quality with database searching
Quality assurance of recombinant proteins is a tough problem, and many analytical techniques are required to verify the primary sequence, modifications, crosslinking, and freedom from undesirable contaminants of the protein product. Although database search was designed for the identification of unknown proteins, it also has a role to play in a QA environment, but we must emphasise that it is for research use only. We are not aware of any regulatory approval for its use in connection with therapeutics (yet!).
Try to avoid searching a sequence database with just one entry, or a very small database where the entries are variants of the same protein. There are no meaningful statistical measures of significance in such searches, making it hard to decide whether a low scoring match is correct or simply a chance peptide molecular mass match. Including common contaminants and the host cell proteome in the search will help give some confidence in the matches even if there are too few for target-decoy validation.
An error tolerant search is ideal for picking up peptides modified by artefacts, such as oxidation or over-alkylation, not to mention non-specific cleavage and the occasional post-translational modification or SNP. For additional tips relevant to QA-type searches, read this recent blog article |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
Top-down and Middle-down Protein Analysis Reveals that Intact and Clipped Human Histones Differ in Post-translational Modification Patterns
Andrey Tvardovskiy, Krzysztof Wrzesinski, Simone Sidoli, Stephen J. Fey, Adelina Rogowska-Wrzesinska and Ole N. Jensen
Molecular & Cellular Proteomics (2015), 14, 3142-3153
Post translational modifications of histones play a major role in regulating chromatin functionality and the concomitant DNA processes. It is thought that not only "classical" histone PTMs, such as methylation, acetylation, and phosphorylation effect the function, but also the proteolytic cleavage of the N-termini known as "histone clipping".
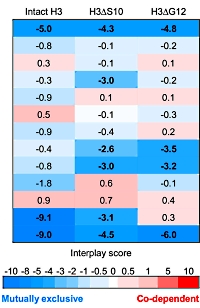
This paper takes on the challenging characterization of the co-existing histone modifications and reveals the relationship between histone clipping and covalent histone PTMs. Using immunoblotting as well as top- and middle-down mass spectrometry methods, the authors showed the presence of clipped histones and the co-existence of various PTMs for H3 histones.
They found that histones H2B and H3 undergo proteolytic processing in primary human hepatocytes and the hepatocellular carcinoma cell line HepG2/C3A. They mapped 212 unique combinatorial PTMs on intact H3 N-terminal tails and 55 combinatorial PTMs on two different clipped H3 N-terminal tails. |
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|