|
To view this email as a web page, click here. |
 |
|
Welcome
If you ever use multiple fragmentation methods in a single acquisition, you should be aware that it is possible to optimise the scoring by specifying the instrument type at the individual scan level. We explain how to do this and what improvement you can expect.
This month's highlighted publication is timely as flu season approaches. The authors look to circumvent the variable efficacy of vaccines.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month describes how Mascot handles ambiguity codes in protein sequences.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
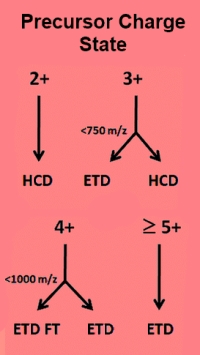
How to get the best from a combined CID+ETD dataset
Today's advanced mass spectrometers are capable of collecting raw data files that contain multiple types of fragmentation. An example would be acquiring CID and ETD scans off each precursor. Since different fragmentation methods usually produce different ion series, the traditional approach has been to search all scans with an instrument definition that is a super-set of all expected ion series.
The down-side of this is that the search space is larger than strictly necessary. The ETD scans will be tested for the presence of b-series ions and the CID scans for c-series ions. Inevitably, this causes some loss of specificity.
Mascot Distiller 2.6 can be configured to output a particular instrument type at scan level according to the scan header in the raw file. When tested against datasets downloaded from PRIDE, we found that searching separately vs combined gave a 13% increase in the number of significant peptide matches and the search ran 20% faster because of the smaller search space.
Go here to read about this in more detail. |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
Phosphoproteomics to Characterize Host Response During Influenza A Virus Infection of Human Macrophages
Sandra Soderholm, Denis E. Kainov, Tiina Ohman, Oxana V. Denisova, Bert Schepens, Evgeny Kulesskiy, Susumu Y. Imanishi, Garry Corthals, Petteri Hintsanen, Tero Aittokallio, Xavier Saelens, Sampsa Matikainen and Tuula A. Nyman
Molecular & Cellular Proteomics (2016), 15, 3203-3219
The influenza A virus season is approaching and many of us will unfortunately be affected. In order to better understand the rapidly mutating virus and its ability to evade our antiviral treatments, the authors have investigated the host cell factors that are exploited by influenza viruses.
Primary human macrophages were infected with influenza A virus to elucidate the intracellular signaling pathways and critical host factors activated upon infection. Using LC/MS/MS and bioinformatics, they identified 1113 human phosphoproteins that showed changes in their phosphorylation status upon IAV infection, implying that the IAV infection effects host protein phosphorylation. Additionally 285 phosphorylation sites in 222 different proteins have not previously been reported.
The infection had a major influence on the phosphorylation profiles of a large number of cyclin-dependent kinase substrates. The authors tested a number of small molecule CDK kinase inhibitors, and showed one to be a potent inhibitor of IAV-associated cellular cytotoxicity. This compound was further shown to rescue infected mice reducing mortality at 10 days from 83% to 0. |
 |
|
|
|
|
|
|
|
Mascot tip of the month
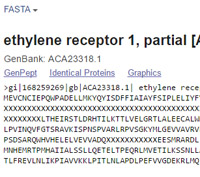
When Mascot tests a peptide containing ambiguous residue codes, it expands and tests all possibilities. For example, a peptide containing a single B will be tested twice, once with N at that position and once with D. Code Z will be tested as E and Q, and the presence of a single X will generate 20 new candidate peptides. To avoid excessive search times, Mascot only expands these possibilities up to a maximum of 3 B or Z residues and 1 X residue per peptide. Above this limit, the weighted average mass will be used, which will usually mean no match.
The abundance of ambiguity codes depends on the database. On average, In NCBI nr, X is present at a level of 1 in 4,000 residues. However, X is not uniformly distributed. It tends to occur in runs where an open reading frame spans a string of N base codes, probably inserted in the course of assembling contigs. The abundance of isolated X residues is less than 1 in 20,000.
On the subject of translation, some databases place an asterisk in the protein sequence to indicate a stop codon in the nucleic acid sequence. This is not a valid single letter code, and will cause Mascot to log an 'illegal character' error during compression. Better to search the nucleic acid sequence directly, because this allows a stop codon to be treated more realistically, as an unconditional gap. Another advantage of working with the original nucleic acid sequence is that, when there is a frame shift, peptide matches in different frames can be kept together. It also enables single base insertions and deletions to be simulated in an error tolerant search. |
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
