|
To view this email as a web page, click here. |
 |
|
Welcome
Some steps in a Mascot search are limited by disk speed and others by processor speed. Understanding these details can be helpful in choosing the right hardware for the job.
This month's highlighted publication demonstrates that an unexpectedly large fraction of cell surface epitopes are the result of peptide splicing, with potential implications for immunotherapies and vaccines.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month explains how to deal with a warning that some of the entries in a sequence database are too long.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
Getting the most out of your Mascot Server hardware
In order to choose the right hardware components for your Mascot Server, it helps to understand the different stages of a search. The overall process flow is this:
- The input file is uploaded
- The spectra are sorted by peptide molecular mass
- The peak list is divided into chunks
- The search itself: the database sequences are digested and fragmented in silico, calculated masses are compared with experimental masses and scores calculated.
- The results are consolidated into the results file
- If requested, Percolator runs to improve the discrimination
- Cache files are created to improve performance for result reports
The main part of the search, when the mass values are calculated and scored, is highly parallel so the processing can be split into a number of independent tasks that run on separate cores of the CPU's. Since the performance bottle neck is normally the CPU, doubling the number of cores used in a search approximately halves the time taken for this step.
The other stages - the sorting and splitting of the peak list, writing the result file, and Percolator post-processing - are currently singly threaded processes, so are not accelerated by using more cores.
Go here to read more about memory requirements and whether it is worth putting certain files on a solid state drive. |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
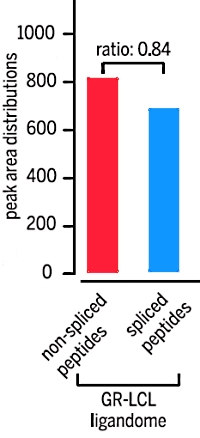
A large fraction of HLA class I ligands are proteasome-generated spliced peptides
Juliane Liepe, Fabio Marino, John Sidney, Anita Jeko, Daniel E. Bunting, Alessandro Sette, Peter M. Kloetzel, Michael P. H. Stumpf, Albert J. R. Heck, Michele Mishto
Science 21 Oct 2016: Vol. 354, pp. 354-358
To identify the presence of pathogens and other maladies, the cells present protein fragments (epitopes) on their surface for targeting by the immune system. These epitopes are created by the action of the proteasome, and then they are displayed by the human leukocyte antigen class 1 (HLA-1) system. The proteasome can also cut proteins and paste different pieces together, forming peptides that do not correspond to the original protein sequences. This process is called proteasome catalyzed peptide splicing and was thought to be rather rare.
In this study, the authors developed a two-dimensional peptide prefractionation strategy followed by a hybrid peptide fragmentation method (electron transfer higher-energy collision dissociation) for peptide identification. The database search employed an adapted target-decoy approach and a vast proteome-wide human spliced peptide database.
This methodology led to the identification of 6592 nonspliced and 3417 spliced peptides 9 to 12 residues in length, which represents 34% of the total of identified antigenic peptides. In contrast, searching these data sets only against the standard Swissprot human proteome database wrongly assigned 655 of the antigenic peptides as nonspliced peptides, while not accounting for the spliced peptides. |
 |
|
|
|
|
|
|
|
Mascot tip of the month
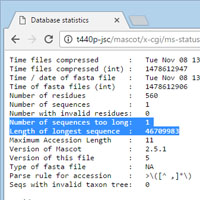
You've added a new database to Mascot; it has been tested and is available for searching. The first time you search it, up at the top of the report, there is a red warning: "3 sequences ignored because length greater than maximum configured".
If you look a little more closely, you'll notice there is a Compression warnings link for the new file in Database Status. Follow this link and you'll see the error messages logged at the time the Fasta file was compressed. You can also follow the statistics link to see how many sequences are too long and the length of the longest.
One way to deal with this would be to increase the value of MaxSequenceLen in the Options section of mascot.dat using the Configuration Editor. In most cases, this is very much the wrong thing to do. The default for MaxSequenceLen is 50,000 residues, comfortably longer that the longest entry in NCBI nr. In fact, you are unlikely to see this warning for a protein database. It tends to occur when trying to add a nucleic acid database containing an assembled genome or some chromosomes. These can be very long - human chromosome 1 is 250 Mb. Trying to search such a long sequence as a single entry makes no sense from the reporting point of view.
It also wastes memory because Mascot has to create tables in memory that scale with the size of the longest entry in the database. Setting MaxSequenceLen to a crazily high value is a guaranteed way to get searches crashing with "Out of memory". Far better to split very long sequences into overlapping chunks, as described here, under Genome Database Example. |
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
