|
To view this email as a web page, click here. |
 |
|
Welcome
Please join our Breakfast Workshops at the upcoming ASMS Conference in Indianapolis, June 5th and 6th.
Protein false discovery rate is a difficult metric to quantify and we discuss some of the factors involved.
This month's highlighted publication shows a new method for enriching and identifying oxidation sites in proteins.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month explains how to add a new element to your local Unimod database.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
Matrix Science Breakfast Workshops at ASMS
We are hosting two Breakfast Workshops at the ASMS meeting in Indianapolis and invite you to join us. Come hear about the latest applications and developments with database searching.
There is no charge for attending these meetings, but advance registration is required. Please go here for details and to register.
Monday 5 June, 7:00 am - 8:00 am
The Future of Peptide Identification by Spectrum Library Searching
presented by Stephen E. Stein, NIST
Integration of spectral library searching into Mascot Server
presented by Matrix Science
Tuesday 6 June, 7:00 am - 8:00 am
The Characterization of Therapeutic Proteins by Top-down and Bottom-up Approaches
presented by Paul W. Brown, Pfizer Worldwide Research and Development
New features in Mascot Server 2.6
presented by Matrix Science
|
 |
|
|
|
|
|
|
|
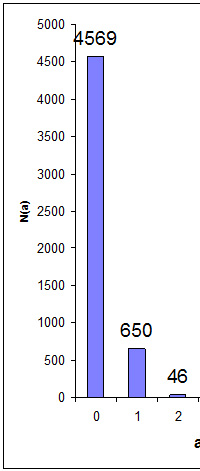
Does Protein FDR Have Any Meaning?
Peptide false discovery rate is easily estimated by searching a decoy database. Estimating the FDR for proteins is a very different matter. What exactly is a false protein? Is it a database entry where all of the peptide match evidence is false? In most cases, we would hope to filter out such proteins by requiring every protein to have significant matches to two or more distinct peptide sequences.
After filtering out 'one-hit wonders', we may still be over-reporting because of the intrinsic ambiguity of protein inference for a set of proteins with shared peptide matches. Whenever we have same-set or near same-set proteins, we either choose one, accepting that we may have picked the wrong one, or report all of them, knowing that they are not all present.
In other words, protein inference in shotgun proteomics is subject to some very serious and fundamental limitations:
- No protein level information In shotgun proteomics, protein level information is discarded in the interests of speed and scale, and protein inference depends on parsimony.
- Low or unknown coverage We cannot assume that a protein with low coverage is a false protein. It could be a true protein that happens to be present at a low level.
- Generic databases Most searches are against the public protein databases, and these will not contain perfectly correct sequences for many of the proteins in the sample. In the absence of the correct sequence, matches are assigned to a set of homologous entries.
- Artefacts from modifications Including an unnecessary modification in a search or omitting a modification that is actually present in the sample can cause a false peptide match that leads to the wrong protein being inferred. The most frequent culprit is deamidation.
Click here to read more about the issues associated with protein inference. |
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
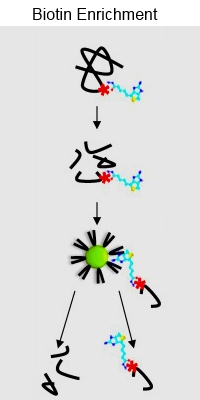
A biotin enrichment strategy identifies novel carbonylated amino acids in proteins from human plasma
Jesper F. Havelund, Katarzyna Wojdyla, Michael J. Davies, Ole N. Jensen, Ian Max Møller, Adelina Rogowska-Wrzesinska
Journal of Proteomics 156 (2017) 40-51
Protein carbonylation is an irreversible protein oxidation and is an important marker of protein damage, related to oxidative stress, disease and ageing. This paper describes a new method for the identification and characterization of 14 different types of carbonylated amino acids in proteins.
The proteins were derivatized with biotin-hydrazide, trypsin digested, and then enriched with monomeric avidin resin. Hot water elution was determined to be the most effective eluting agent, outperforming other approaches. Additionally, the authors developed a script to specifically detect and remove biotin ions from spectra, improving scores for most peptides.
Using this method, they identified 125 carbonylated residues in oxidized bovine serum albumin and assigned 133 carbonylated sites in 36 proteins in native human plasma protein samples. They also detected 10 previously undetected types of carbonylated amino acids in proteins.
|
 |
|
|
|
|
|
|
|
Mascot tip of the month
Unimod includes most of the elements you are likely to encounter in protein work, but it is possible you might need to add one. The browser-based configuration editor doesn't include a module for elements, so you have to edit one of the configuration files. This is easy enough, but if you are using Mascot 2.5 or earlier, any changes to unimod.xml (or master.xml in version 2.5) will be lost when you download a new unimod.xml file from the Unimod web site. Best to
ask Unimod to add the missing element to the public database.
In Mascot 2.6, local changes to elements and molecules are stored in a separate file, so that nothing is lost when you download an update.
To illustrate the procedure, let's assume we want to add an entry for Platinum. WebElements is a convenient site to get the mass values. Click on the appropriate position in the periodic table to load the summary page for Platinum. The average mass is listed at the top of the page (Relative atomic mass 195.084). For the monoisotopic mass, you have to look further down the page, under isotopes. Remember that monoisotopic mass is the mass of the most abundant natural isotope, not the lightest, so the correct value for Platinum is 194.964766
In Mascot 2.6 and later, open mascot\config\unimod\usermod.xml in a plain text editor.
Add this row to the elements section, immediately before the closing tag (</umod:elements>)
<umod:elem avge_mass="195.084" full_name="Platinum" mono_mass="194.964766" title="Pt"/>
Add these rows to the mod_bricks section, immediately before the closing tag (</umod:mod_bricks>)
<umod:brick avge_mass="195.084" full_name="Platinum" mono_mass="194.964766" title="Pt">
<umod:element number="1" symbol="Pt"/>
</umod:brick>
After saving these changes, you should find that Platinum can now be selected for new modifications in the configuration editor. Reward yourself by visiting the WebElements shop and buying an item of periodic table-themed clothing. |
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
