|
To view this email as a web page, click here. |
 |
|
Welcome
You can readily create a custom spectral library to match contaminant spectra
without increasing the search space for proteins of interest.
This month's highlighted publication demonstrates a new enrichment method that enabled thousands
of non-canonical phosphopeptides to be identified.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month describes how to exclude a class of modifications, such as isotopic labels, from an error tolerant search.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
Spectral library for contaminants
A custom spectral library can be used to identify spectra from contaminant proteins without increasing the search space for proteins of interest. This is particularly useful for sequencing-grade trypsin, which is modified by methylation or acetylation of the lysines, creating a large number of modified non-specific peptides that are missed by typical search strategies.
It would be ideal if you could download a ready-made library of all common contaminants, but variation in artefactual modifications due to variation in experimental protocols means you really need to create your own. This is not difficult, and Mascot 2.6 includes all the necessary tools.
Here is a detailed, step-by-step procedure to build your own contaminants library. You can run searches for additional contaminants at any later time and add these to an existing library. To use the library in a search, you simply select it alongside a suitable Fasta for your target proteins.
|
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
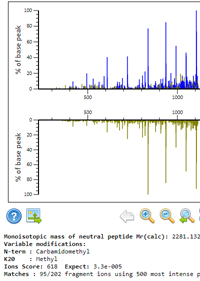
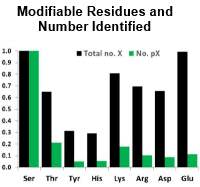
Extensive non-canonical phosphorylation in human cells revealed using strong-anion exchange-mediated phosphoproteomics
Gemma Hardman, Simon Perkins, Zheng Ruan, Natarajan Kannan, Philip Brownridge, Dominic P Byrne, Patrick A. Eyers, Andrew R. Jones and Claire E. Eyers
bioRxiv posted 13 October 2017, 202820
Protein phosphorylation is an essential PTM, and there are many well-established workflows to determine the localization of phosphorylation on peptides containing serine, threonine, and tyrosine residues. Based on evidence suggesting that histidine phosphorylation also plays a role in human biology, the authors set out to develop a method to find these sites of non-canonical phosphorylation.
The phosphoramidate bond in phosphohistidine is susceptible to hydrolysis under the traditional acidic conditions used for phosphopeptide analysis. To avoid this, they developed an enrichment strategy based on strong anion exchange chromatography, providing enrichment of acid-labile phosphopeptides at or near neutral pH.
They were able to extend this approach to other labile phosphosites and in a HeLa cell lysate identified ~300 pHis-containing peptides and 2740 peptides with sites of phosphorylation on Arg, Lys, Asp and Glu.
|
 |
|
|
|
|
|
|
|
Mascot tip of the month
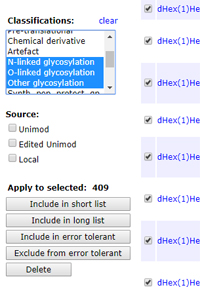
By default, an error tolerant search tests every modification in the local Unimod file. Many of these modifications, such as isotopic labels, cannot possibly be present in the sample. The argument for including them is that, if you get a strong match to an important protein, or if the modification comes up many times, it might be worth trying to figure out what's going on. A valid mass delta has been found, it's just the description that is wrong.
On the other hand, Unimod gets larger every year. If the first pass search identifies a large number of proteins, the second pass search can take a long time. Excluding a class of modifications that is not expected to be present is an easy way to speed things up. Glycosylation is a good example. This accounts for some 400 modifications, mostly larger than 500 Da.
Provided you are running Mascot 2.5 or later, excluding a class of modifications is very easy. From your Mascot home page, follow the link for Configuration Editor, then Modifications. Filter the list by selecting one or more classifications, then set Page size to All. Check the box next to the title column header to select all listed modifications, then choose Exclude from error tolerant.
|
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
