|
To view this email as a web page, click here. |
 |
|
Welcome
We highlight some of the basic, but often overlooked, considerations for a successful search.
This month's highlighted publication elucidates the largest set of CSF proteins identified by a rigorous analysis of human cerebrospinal fluid.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month concerns troubleshooting corrupt XML data files.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
Common mistakes you have never made …
We have looked at some of the reasons why searches fail to give good results on our public web site. The causes are often very simple, and it may be helpful to review a few of them here.
- Choose your database wisely Mascot score does not change with database size, but using an unnecessarily large database or too many databases increases the size of the search space leading to higher significance thresholds and fewer significant matches.
- Include a contaminants database If you search a single organism database or use a taxonomy filter with a comprehensive database, it's important to include a contaminants database. Many spectra are likely to be from contaminants, such as trypsin. If the correct protein is not included in the search, peptide matches from contaminants may lead to the wrong protein being inferred.
- Don't use an under-represented taxonomy Some species are poorly represented, even in a very large database. For example, Hystricidae (Old World porcupines) has only 181 protein sequences in NCBIprot. In such cases, move up the taxonomy tree to a better populated level, such as Rodentia, since many proteins from porcupines will have extensive homology with their less prickly cousins.
- Insufficient or impossible masses A decent MS/MS spectrum will have at least one peak for each residue. If you have a spectrum with just two or three peaks, you cannot expect to get a significant score. For a PMF of a tryptic digest, you need a reasonable number of peaks in the mass range that is typical for tryptic peptides - between 1000 and 3500 Da. A very common error is to submit a PMF search with a single mass value. It isn’t possible to get a significant protein match from a single mass value, however good the accuracy, because any given residue composition is likely to occur in many different proteins.
Go here to read more about getting the best from your searches.
|
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
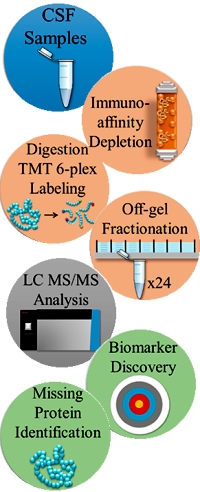
Deep Dive on the Proteome of Human Cerebrospinal Fluid: A Valuable Data Resource for Biomarker Discovery and Missing Protein Identification
Charlotte Macron, Lydie Lane, Antonio Nunez Galindo, and Loic Dayon
J. Proteome Res., Article ASAP, published online August 20, 2018
Human cerebrospinal fluid is a window into the brain and a useful source for neurobiological biomarkers for clinical studies. In this paper, the authors have undertaken a comprehensive analysis of a commercial pool of "normal" CSF samples to deeply elucidate the proteome.
The pooled CSF sample was depleted of abundant proteins, labeled with tandem mass tags (TMT), and fractionated in 24 fractions using off-gel electrophoresis. Each fraction was analyzed independently with RP−LC tandem MS (MS/MS).
This shotgun proteomic analysis identified 20,689 peptides mapping on 3379 proteins, and is the largest CSF proteome published so far. Among the CSF proteins identified, 34% correspond to genes whose transcripts are highly expressed in brain according to the Human Protein Atlas. Additionally 26 "missing proteins", predicted by genomic or transcriptomic analyses, were also identified.
|
 |
|
|
|
|
|
|
|
Mascot Tip
If there is a problem with an XML format raw file or peak list, the error message you get from the XML validator in Mascot Server or Mascot Distiller may not be very helpful. A typical message is "The key for identity constraint of element 'mzML' is not found."
This tip describes how to get more verbose error information. To keep the example brief, we assume the platform is Windows and the file format is mzML, but the toolkit is also available for Linux and the same approach can be used for other XML files
- Download the XMLStarlet toolkit from Sourceforge (green button for Windows version)
- Unpack to a suitable directory
- Download the mzML schema to the same directory. There are two files: mzML1.1.0.xsd is the main schema and mzML1.1.1_idx.xsd is also required for files which include the optional index.
- Enter the following in a command window, with paths and filenames changed appropriately. Note that everything from xml.exe onwards should be entered as a single line.
cd "C:\scratch\xmlstarlet-1.6.1"
xml.exe val --err --xsd mzML1.1.1_idx.xsd ..\data\problem.mzML 2> ..\data\errors.txt
The error messages written to errors.txt may be sufficient for you to figure out the problem and fix the XML file in a text editor. If not, please attach errors.txt to your message when you contact support@matrixscience.com.
|
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
