|
To view this email as a web page, click here. |
 |
|
Welcome
We have some tips and observations concerning the Mascot Server node in Proteome Discoverer.
This month's highlighted publication illustrates how peak intensities can give more accurate quantitation results than peak areas.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month describes the different types of neutral loss that can be associated with modifications.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
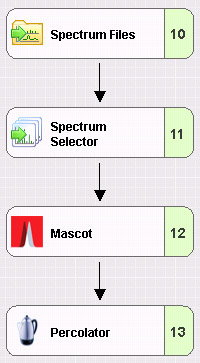
Mascot Server workflows in Proteome Discoverer
For those of you that use Thermo's Proteome Discoverer (PD) as your primary user interface for database searching, we have a few tips and observations:
- Peak list size
PD splits large peak lists into separate searches and transparently merges the results back together. If you want to view the search results using Mascot reports, and perform repeat searches, you can still do this by creating a combined result report, as described in our May 2017 newsletter.
- Missing functionality
There are a few features in Mascot Server 2.6 that are not supported by the PD Mascot node, such as multiple precursor masses for chimeric spectra, error tolerant searches, and searching combinations of Fasta files and spectral libraries. To access these features for data processed through PD, repeat the search via the Mascot Server search log.
- Target Decoy PSM Validator
For large search spaces, such as no enzyme searches, searches with many variable modifications, or very large databases, the Target Decoy PSM Validator can be wildly over conservative. For better sensitivity, use the Percolator node instead.
Go here to discover more tips and observations concerning the Mascot Server node in Proteome Discoverer.
|
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
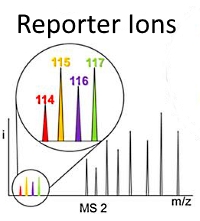
Sum of peak intensities outperforms peak area integration in iTRAQ protein expression measurement by LC-MS/MS using a TripleTOF 5600+ platform
Bastien Burat, Julien Gonzalez, Francois-Ludovic Sauvage, Hassan Aouad, Helene Arnion, Emilie Pinault, Pierre Marquet and Marie Essig
Bioscience Reports, 39(6) BSR20190904 (2019)
The authors investigated the quantitative accuracy of using peak areas versus peak intensities for 4-plex iTRAQ experiments of calcineurin inhibition on renal proximal tubular cells. They built an automated data processing algorithm called Customizable iTRAQ Ratio Calculator (CiR-C) to refine critical parameters related to peptide confidence and selection.
The data workflow encompassed Mascot Server for peptide and protein identification, jTRAQx software for computation of summed peak intensities at the peptide level, and the CiR-C algorithm for data integration and final protein quantitation. In comparison with the peak area workflow, the summed peak intensity approach had superior linear correlation with Western blot quantitation.
|
 |
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|