|
To view this email as a web page, click here. |
 |
|
Welcome
Studying complex metaproteomics samples can lead to many shared peptides, but clustering can help decipher the proteins.
This month's highlighted publication shows a new approach to examine crosstalk of modifications in synaptic proteins.
If you have a recent publication that you would like us to consider for an upcoming Newsletter, please
send us a PDF or a URL.
Mascot tip of the month explains how a variable modification can displace a fixed one in a Mascot search.
Please have a read and feel free to contact us if you have any comments or questions. |
|
|
|
|
|
 |
|
|
|
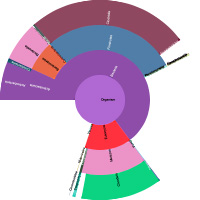
Leverage shared peptides for complex samples
Protein identification is particularly challenging for the complex mixtures encountered in metaproteomics studies and environmental samples. Since the proteins from different species cannot be separated during sample processing, shotgun protein sequencing is done on the mixture proteome.
It is common to get a match to two similar protein sequences because the analyte is partly represented by both sequences, especially in metaproteomics, where nearly all sequence databases are approximate or incomplete. Additionally, the sample is likely to contain multiple similar bacterial strains as well as closely related species, which means shared peptides are inherent to the data.
The key is to use all the available peptide matches for protein inference by utilizing protein clustering. In the Mascot Protein Family Summary, proteins are clustered into families based on shared peptides, and all top-level proteins in the family have at least one unique, significant peptide match. You will see homologous proteins from different species pulled together.
For a human gut microbiome study, we illustrate how the clustering can help you unravel these complex samples. Go here to read more.
|
 |
|
|
|
|
|
|
|
Featured publication using Mascot
Here we highlight a recent interesting and important publication that employs Mascot for protein identification, quantitation, or characterization. If you would like one of your papers highlighted here please send us a PDF or a URL.
|
|
|
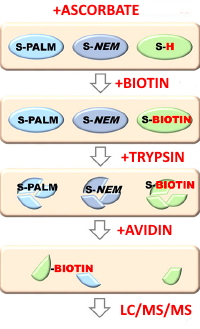
Stress-induced Changes in the S-palmitoylation and S-nitrosylation of Synaptic Proteins
Monika Zareba-Koziol, Anna Bartkowiak-Kaczmarek, Izabela Figiel, Adam Krzystyniak, Tomasz Wojtowicz, Monika Bijata, and Jakub Wlodarczyk
Molecular & Cellular Proteomics, 18 1916-1938 (2019)
The authors developed a new approach to investigate posttranslational modifications of synaptic proteins that are differentially affected by stress. Specifically, they examined the crosstalk between S-palmitoylation and S-nitrosylation, since an imbalance in the dynamics of these PTM's is associated with neurodegenerative disorders such as Alzheimer's disease, Huntington's disease, schizophrenia, and Parkinson's disease.
Their method, Palmitoylation And Nitrosylation Interplay Monitoring (PANIMoni), involves selectivity reducing the synaptic proteins with either hydroxylamine (for PALM detection) or ascorbate (for NO detection), biotin labelling of the newly-formed thiol sites, trypsinolysis, affinity enrichment, and then LC-MS/MS for ID and label-free quantitation.
They identified 813 S-PALM and 620 S-NO cysteine sites on 465 and 360 proteins, respectively. Of these proteins, 122 were differentially expressed in the stressed mouse model, showing S-palmitoylation and S-nitrosylation crosstalk in the regulation of receptors, scaffolding proteins, regulatory proteins and cytoskeletal components.
|
 |
|
|
|
|
|
|
|
Mascot Tip
A simple tip, this month, but one that comes up again and again in technical support.
A standard search can only include a single fixed modification for each specificity. For example, you can have Carbamidomethyl (C) or Propionamide (C) as fixed modifications, but not both at the same time. Maybe your experimental protocol means that either can be expected, but never an unmodified Cys. If so, you can choose one as fixed and the other as variable. The rule is that a variable mod can displace a fixed one, so this keeps the search space as small as possible, because unmodified Cys is never considered. If a peptide had multiple Cys, then Mascot would look for matches with all possible arrangements of the mods, not just all Carbamidomethyl or all Propionamide.
The main time that you need greater flexibility with modifications is for an isotopic labelling experiment, where additional options can be specified as part of a quantitation method, as described in our June 2019 newsletter.
|

|
|
|
|
|
|
|
|
About Matrix Science
Matrix Science is a provider of bioinformatics tools to proteomics researchers and scientists, enabling the rapid, confident identification and quantitation of proteins. Mascot software products fully support data from mass spectrometry instruments made by Agilent, Bruker, Sciex, Shimadzu, Thermo Scientific, and Waters.
Please contact us or one of our marketing partners for more information on how you can power your proteomics with Mascot.
|
 |
|
|
|
|
