Peak picking intact crosslink spectra with Mascot Distiller
There are several important factors to take into account when carrying out peak picking for an intact crosslinked dataset. You’ll typically be dealing with higher charge state precursors to handle the larger masses of the linked peptides, and the MS/MS spectra are inherently complex, chimeric spectra with fragments from the alpha and beta peptides.
This page illustrates the effects of peak picking settings in Mascot Distiller. The example data comes from PRIDE project PXD014337, where Beveridge et al. (Nature Communications 11, 742 (2020)) created a synthetic peptide library for benchmarking crosslinking-mass spectrometry search engines for proteins and protein complexes.
Fragment ion charge states
Mascot Server only searches for 1+ and 2+ fragment ion series, but if you have higher charge state precursors, many of the fragment ions generated will similarly have higher charge states. At best, they can’t be matched; at worst, they might result in false positive matches.
With these types of datasets, you should either de-charge the fragment ion peaks to MH+ or supply the peak charge states in the peak list. Mascot Distiller can do this, but if your MS/MS scans are saved as centroids, rather than profile, you need to use the uncentroiding options first.
The example data set was acquired with a Thermo Orbitrap instrument. Peak picking was carried out using the following settings:
- default.ThermoXcalibur.opt, which simply take the centroid values from the raw file without determining charge state
- prof_prof.ThermoXcalbur.opt, which uncentroid the MS/MS scans at a resolution of 600 ppDa
The peaklists were then searched using Mascot Server. Where MS/MS scans had been uncentroided, the option to output fragment ion mass values as MH+ was selected in Mascot Distiller, under the “Peak list format” options in the preferences dialog. Results are presented in table 1 below:
| Processing options | Uncentroiding points per Da | Decharged fragment ions? | Time taken (mm:ss) | Significant CSMs (p<0.05) |
|---|---|---|---|---|
| default.ThermoXcalibur.opt | N/A | No | 00:14 | 655 |
| prof_prof.ThermoXcalibur.opt | 600 | Yes | 12:08 | 1143 |
Table 1: Comparison of processing time and significant cross-linked spectrum match results (CSMs) using default and the prof_prof processing options.
The results from default.ThermoXcalibur.opt are significantly worse. This is because there are many spectra containing fragment ions with charge states in excess of 2+, which will not be matched by Mascot. Figure 1 below shows a crosslinked match from a 4+ precursor using the default.ThermoXcalibur.opt and the same match using the prof_prof.ThermoXcalibur.opt settings:
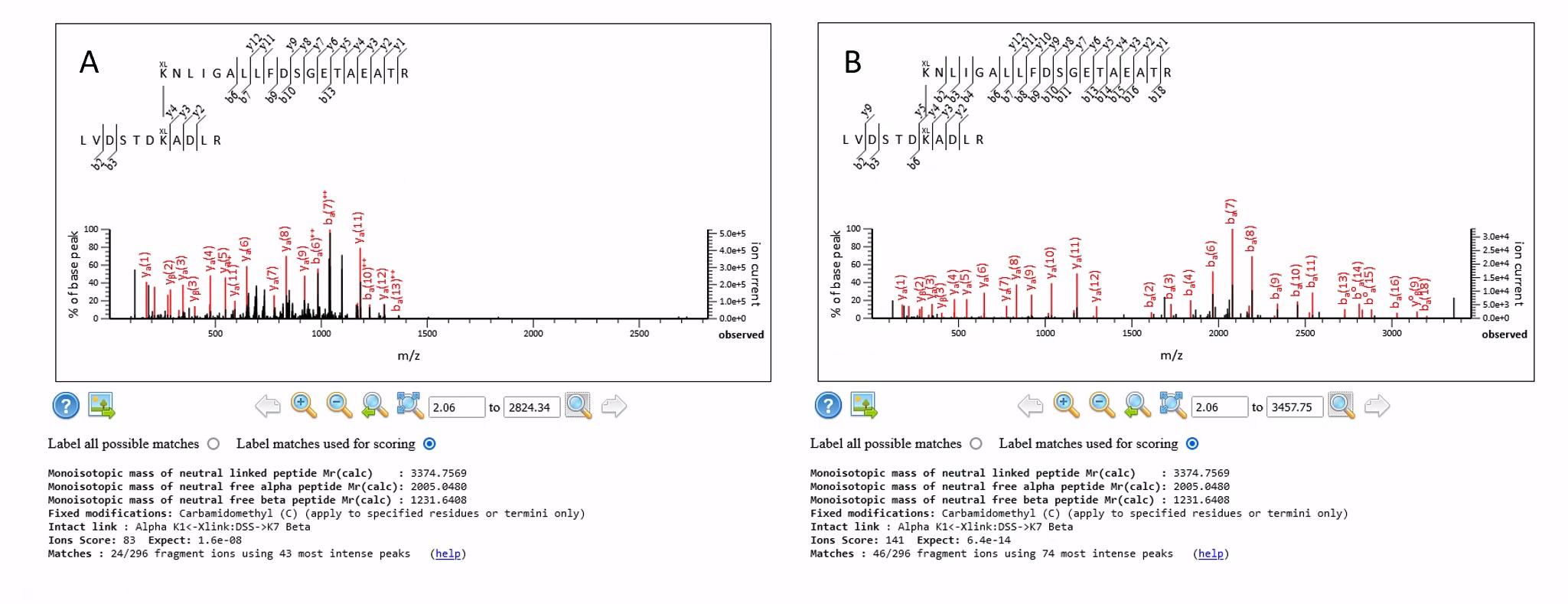 Figure 1 The same match where the data were processed using A) default.ThermoXcalibur.opt and B) prof_prof.ThermoXcalibur.opt and then decharged
Figure 1 The same match where the data were processed using A) default.ThermoXcalibur.opt and B) prof_prof.ThermoXcalibur.opt and then decharged
Both are strong matches, but there are a reasonable number of 3+ fragment ions (and a few 4+), so the decharged peak list from prof_prof.ThermoXcalibur.opt is giving increased coverage of both the alpha and beta peptides. The higher charge state fragments can now be matched, increasing the coverage of the fragment ions from 24 to 46 and improving the score from 83 to 141.
Uncentroiding resolution
While the prof_prof.ThermoXcalibur.opt settings out-performed the default options, this comes at a heavy cost in processing time.
Decreasing the uncentroiding setting in the prof_prof option to 200 or 400 ppDa can still give good results while improving processing time. Results are summarised in table 2 below:
| Uncentroiding ppDa | Time taken (mm:ss) | Significant CSMs (p<0.05) |
|---|---|---|
| 200 | 02:33 | 1139 |
| 400 | 06:25 | 1141 |
| 600 | 12:08 | 1143 |
Table 2: Results from adjusting the prof_prof.ThermoXcalibur.opt processing settings to use 200 and 400 points per Da to uncentroid the MS/MS scans
As you can see, using either 200 or 400 ppDa both yield results close to the default of 600 ppDa, while taking approximately 20% and 50% the processing time, respectively. The reasons for these differences are the same: uncentroiding to a higher resolution improves peak picking and reduces the risk of nearby adjacent centroids being merged into a single uncentroided peak. Reducing the uncentroiding resolution to 400ppDa seems to be a reasonable trade off between speed of processing and the quality of the results. Of course, if your data are saved as profile, rather than centroids, then this is not a trade-off you need to make.
Signal to noise
Good signal to noise will generally result in a higher confidence match. MS/MS spectra from intact crosslinked peptides are inherently chimeric, making the signal to noise of the peaklists even more important.
Figure 1 above shows that carrying out peak picking in Mascot Distiller instead of simply taking the centroids provided by the instrument is giving cleaner peaklists with better signal to noise. However, it’s also clear that the default signal to noise cut off of 1 may be too low for these data, so we reprocessed the peaklists with an increased value of 2 for the “Minimum signal to noise (S/N)” setting on the MS/MS Peak Picking options. Results are summarised in table 3:
| Uncentroiding ppDa | Minimum signal to noise | Time taken (mm:ss) | Significant CSMs (p<0.05) |
|---|---|---|---|
| 400 | 1 | 06:25 | 1141 |
| 400 | 2 | 05:24 | 1152 |
Table 3: Results from the adjusted prof_prof.ThermoXcalibur.opt processing settings using 400 ppDa for uncentroiding and a minimum signal to noise of either 1 or 2 for MS/MS peak picking.
For these data, increasing the signal to noise threshold has yielded an additional 11 CSMs, and reduced the processing time by 1/6th. What threshold to use on your own data will depend on your instrument. Setting too high a threshold will result in correct fragment ions being excluded from the peaklists, resulting in worse database search results. Run a standard sample through your workflow, adjust the processing options to give them best combination of results and processing time for your data, and then use those as your default settings for processing with Distiller.
The example above is from a Thermo instrument, where MS/MS scans are routinely saved as centroids. Many non-Thermo instruments default to collecting data in profile mode, so you won’t need to uncentroid. The points about decharging the fragment ions and adjusting the signal to noise thresholds apply to all instruments.