Reviewing error tolerant results
This example uses a small error tolerant search to illustrate different aspects of an automatic error tolerant search.
Protein hits
It is important to recognise that only the matches from the standard, first pass search provide evidence for the identity of a protein. The additional matches found in the error tolerant, second pass search are valuable because they are the most likely assignments of the spectra. Occasionally, an additional match will provide useful biological information, such as distinguishing between two isoforms. If the same modification shows up many times, this may indicate an experimental artefact that needs to be eliminated or, at least, selected as a variable modification for standard searches.
Nevertheless, these additional matches have been obtained by selecting a small number of database entries and beating them into submission with non-specificity, substitutions and a long list of modifications, so should be viewed with caution.
The second pass matches do not contribute to the protein score. If the query also has a lower scoring match to the same protein in the first pass search, this contributes, so that the protein scores are identical to those that would be obtained in a standard, single pass search.
Score thresholds
The target FDR is applied independently to the results from the second pass, which means that the significance thresholds for the two passes may be very different. Since the target is based on PSM counts, if it can be achieved for the results from both passes, then it will also be true for the combined results. If it is not possible to get within a factor of 2 of the target, a warning will appear in the report.
Queries that get significant matches in the first pass search don’t have error tolerant results. Queries that failed to get significant matches in the first pass go forward to the second pass, where they are searched against the selected entries. Statistics for the second pass matches are based on the total number of trials from both passes. This could mean that a much higher score is required to get a significant match in the second pass than in the first.
Alkaline phosphatase
For example, click on this thumbnail image to load an example of the results from an automatic error tolerant search with decoy. Scroll down to hit 2, Alkaline phosphatase.
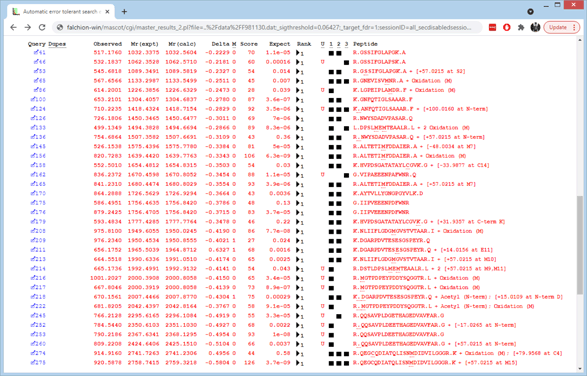
In some cases, the additional match is the result of non-specific cleavage, such as queries 133 and 162. If the error tolerant match was found by introducing a modification or a sequence change, the mass delta and its location are given at the end of the row, in square brackets. When the mouse rests over the mass delta hyperlink, all the known assignments of this delta are displayed in a pop-up.
Take a look at query 260. The mass tolerance for this search was fairly wide, ±0.8 Da, so the observed mass difference could correspond to either carbamidomethylation or carboxymethylation at the N-terminus. Since this sample was alkylated with iodoacetamide, we would choose carbamidomethylation as the more likely suspect, especially as this brings the error on the precursor mass into line with the general trend, whereas carboxymethylation would give an error of +0.5 Da. The assignment to carbamidomethylation is also very believable, because this is a known artefact of over-alkylation. The same modification is found for other queries. Another easily believable assignment is pyro-Glu for the match to query 252.
In other cases, the match may be good, but the assignment is not believable. For example, look at query 218, which has a mass difference of 15.0 Da on the N-term D, assigned to Hydroxamic_acid (DE). If you look this up in Unimod, it is described as an artefact of exposure to hydroxylamine. This is possible, but note that the amino terminus also carries an Acetyl, mass 42.0, which was included in the search as a variable modification. The sum of these two mods is 57.0, which happens to be the mass of N-term carbamidomethylation, an altogether more likely explanation.
Always check the alternative matches that are displayed if you expand the twisty in the rank column or click on the query number to load a Peptide View report. It is common to get multiple matches with similar scores, and the best match may be an unlikely modification, while a match with a slightly lower score has a more credible explanation. Query 124 provides a good example. The displayed match has a delta corresponding to succinylation and the peptide sequence found in family member 2.3. A match with the same score is obtained from the peptide sequence found in family member 2.1 plus a delta of 114.0, which corresponds to double carbamidomethylation – a more likely modification.