Identifying most common trypsin modifications
The Journal of Proteome Research has a paper from the Medical University of Graz concerning the importance of correctly identifying spectra from contaminant proteins. In particular, trypsin autolysis peptides.
The authors point out that sequencing grade trypsin is modified by methylation or acetylation of the lysines, to inhibit autolysis. Unless these variable modifications are selected in a search, simply including a contaminants database will not be sufficient to catch all trypsin autolysis peptides. As part of their study, data were acquired using an LTQ-Orbitrap Velos from a yeast cell lysate digested with Promega trypsin (Data Set 1 in the paper). The raw data is available on PRIDE.
Example search
This example uses three raw files from PRIDE project PXD002726. Files were processed into a single merged peak list using Mascot Distiller.
The search results:
Search parameters can be inspected by expanding the relevant section in the report header. It is also important to consider the composition of the Unimod database. In this case, it was the latest update, but modifications classified as isotopic labels and those with deltas greater than 1000 Da were excluded from the error tolerant search. This reduced the number of modifications from 1499 to 1045.
Expand the Sensitivity and FDR section to see that the sensitivity at 1% FDR for PSMs was 4279. The protein FDR doesn’t look very pretty, but this is a consequence of the 42 decoy PSMs being scattered randomly across 20 decoy proteins. If we increase the Min. number of sig. unique sequences from 1 to 2 and choose Format, the protein FDR drops to a more satisfactory 0%.
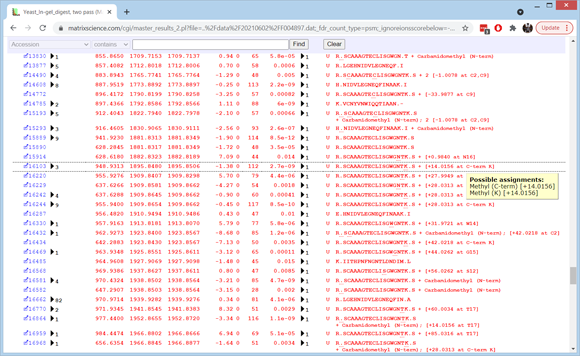
If you want to see how many additional matches were obtained in the second pass search, make a note of the PSM count (4279) then set Error tolerant matches to None and choose Format. The count drops to 2972, so there were 1307 new matches at 1% FDR. Set Error tolerant matches back to Reliable and choose Format again. Expand the Modification statistics section of the header to get an overview of where the error tolerant matches come from. Non-specific cleavage is up near the top of the list, along with deamidated, ethyl, methyl, and guanidinyl. Most of the deamidation and guanidination will be artefactual. Much or all of the ethyl is really dimethyl, which is unusually abundant because trypsin is methylated to reduce autolysis.
To see details of individual matches for the top 10 protein families, choose Expand all at the bottom of the page. The first family are human keratin contaminants. Scroll down to the matches to R.TSQNSELNNMQDLVEDYK.K. There is a first pass match to the oxidised peptide for query 18970 and a second pass match to the oxidised and deamidated peptide for query 18999. Both have a score of 99 but the first pass match has an expect value that is lower by a factor of 300, reflecting the smaller search space. If you hold the cursor over the value in the rank column, you can see that the score threshold is 18 for the first pass and 39 for the combined passes.
Although this example is dominated by artefactual modifications, and it is hard to spot any post-translational modifications, it serves to demonstrate how the new features work to provide confidence in the matches and modifications being reported. For more detailed information, refer to the help page for error tolerant searches.