O-fucosylated CID spectra
O-linked fucose is easily lost in CID. Swearingen et al. discussed this in the Journal of Proteome Research in the context of identifying O-fucosylated thrombospondin type 1 repeats (TSRs) in Plasmodium parasites using database searching. The main problem is, the O-glycosidic bond is weaker than the peptide backbone. Collision energies typical for peptide fragmentation cause it to break, leaving behind mostly unmodified fragments. A related issue is that the O-linked fucose itself is sometimes modified by an equally labile glucose, while tryptophan can carry a stable C-mannose isobaric to glucose.
Mascot has no problems with O-fucosylation, as long as its neutral losses are configured correctly.
Hex, dHex and dHex(1)Hex(1)
If a variable modification has multiple scoring neutral losses, all are tried during the database search. The one giving the highest score is chosen and the rest are treated as satellite NLs. Satellite peaks are treated as “not noise”, which improves the score. The net effect is, Mascot can match fragments from both the unmodified peptide and the O-fucosylated peptide in the same spectrum.
Suitable modifications already exist in Unimod. C-mannose is modelled as a hexose, Hex (W). O-fucosylation is available as deoxy hexose, dHex (ST), while the O-linked fucose-glucose disaccharide is dHex(1)Hex(1) (ST). All the Unimod glycan definitions take their labile nature into account: most specificities have a neutral loss equal to the modification delta.
Swearingen et al. point out that C-mannosylated tryptophan can suffer from cross-ring cleavage, which is a neutral loss of 120Da (C4H8O4). This is currently missing from the “official” Hex (W) definition, but it’s easy to add in your local Mascot Server.
- Go to the Configuration Editor and open Modifications.
- Navigate to the Hex definition.
- Click on “Make editable”.
- In the Specificity tab, click Show Details next to the W specificity.
- Click New Neutral Loss and type in C(4) H(8) O(4). Ensure the NL type is Scoring.
- Save.
It’s important that Hex (W) also has a zero neutral loss, because this models the intact C-mannose. In the editor, zero NL appears as a blank neutral loss:
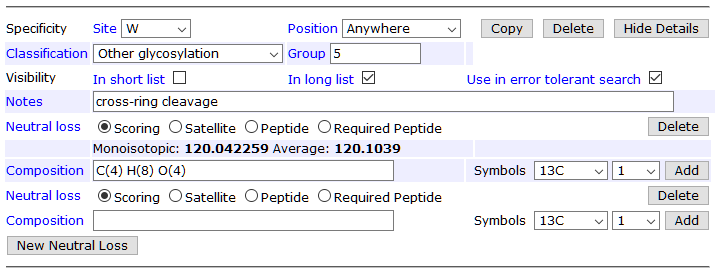
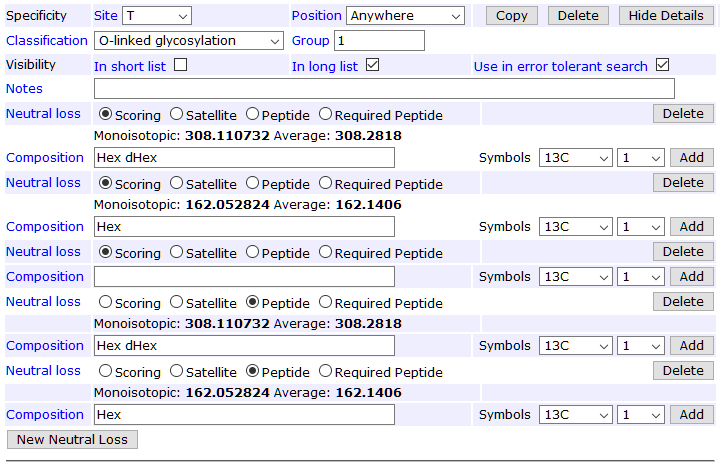
The other missing piece is allowing the loss of a hexose from dHex(1)Hex(1), which models the loss of glucose. Additionally, it’s beneficial to define two peptide neutral losses corresponding to the two scoring neutral losses. At the collision energies used by Swearingen et al., nearly all O-fucosylated MS/MS spectra have a dominant precursor peak for the unmodified peptide. A peptide NL peak, if present, is treated as “not noise” like satellite NL peaks.
The screenshot below shows the full neutral loss definition for specificity T; the list is the same for S.
Tip: You can see which modifications have been edited locally by ticking Edited Unimod under the Source section in the left-hand bar of the main Modifications page.
Example search
This example uses the recombinant P. falciparum thrombospondin-related anonymous protein (PfTRAP) data set from PeptideAtlas (PASS01201) as mzML (2017-10-12_KES_PfTRAP_FTCID_TSRtargeted.mzML). The mzML file contains centroided data, so can be searched directly with Mascot. The basic procedure for optimizing search parameters yields:
- Database: use SwissProt with Plasmodium falciparum taxonomy and cRAP (there are not enough protein sequences to do a meaningful decoy search)
- Precursor tolerance: 10ppm (same as in the paper)
- MS/MS tolerance: 0.02Da (ditto)
- Fixed modifications: Carbamidomethyl (C) (ditto)
- Variable modifications: Oxidation (HW), dHex (ST), Hex (W), dHex(1)Hex(1) (ST)
- Enzyme: Trypsin/P with 2 missed cleavages
- Instrument: ESI-TRAP
This is a targeted DDA data set, whose inclusion list contains the masses for the unmodified TASCGVWDEWSPCSVTCGK plus different numbers of O-fucosylation on serine/threonine and C-mannosylation on tryptophan. In a larger data set, it’s possible that other sites are C-mannosylated as well. An error tolerant search is the best way to discover possible modifications and their specificities.
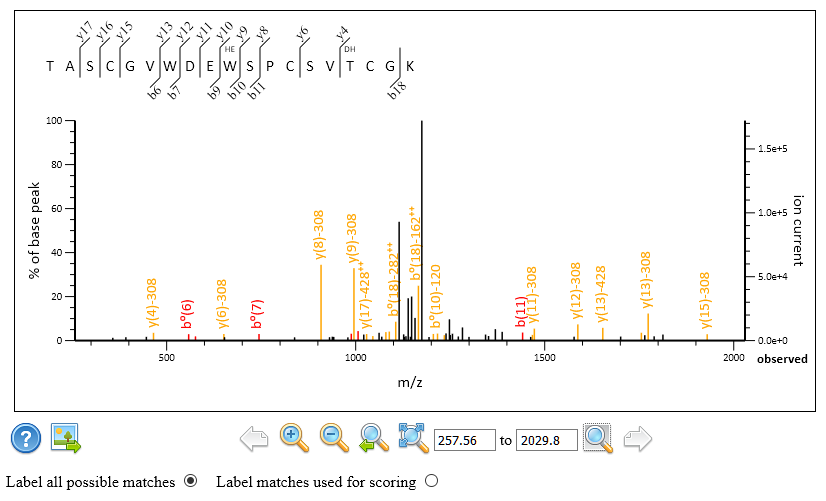
The results are very similar to what Swearingen et al. reported. In particular, scan 9260 of figure 4 of the paper gets the same match to TASCGVWDEWSPCSVTCGK with one Hex (W) and one dHex(1)Hex(1). You can see in Peptide View that Mascot chose the unmodified fragments (NL 308Da) as the rank 1 match. The tallest peak in the spectrum is the peptide NL peak corresponding to the same loss of dHex(1)Hex(1). In the screenshot below, all possible peaks are annotated, not just those used for scoring.
However, look at the site assignment:
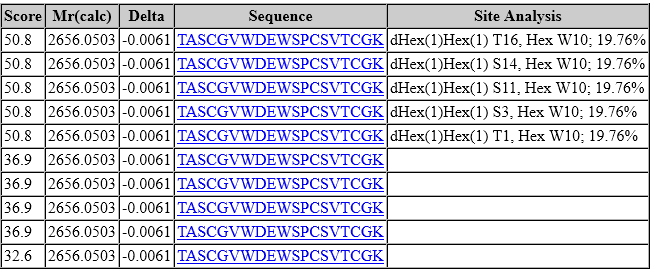
In the top 5 ranks, tryptophan (W10) carries a C-mannose, but there is complete ambiguity among the five different possible sites for dHex(1)Hex(1). In fact, this is true for all matches to peptides carrying dHex(1)Hex(1). The lower-scoring matches (score 36.9) have an oxidised tryptophan (W10) combined with permutations of two fucoses (one dHex(1)Hex(1) and one dHex). It’s clear W10 is modified and very likely C-mannosylated. However, as dHex(1)Hex(1) is lost quantitatively or near-quantitatively, peaks selected for scoring contain no information for site localisation.
Finally, the authors highlight a match to scan 9201, which gets the incorrect modification assignment 2xHex (W) using a third-party software package. This happens because the software is allowed to consider neutral loss of O-linked fucose independent of modification assignment, even though Hex (W) cannot lose a fucose from a tryptophan. Interestingly, Mascot finds the correct match. The incorrect mod assignment is impossible, because the neutral loss is part of the definition of dHex(1)Hex(1).