Does your search engine show the evidence?
You’ve submitted a protein sequence database search and start looking at the results. Why did the search engine identify that protein? What is the peptide evidence? Which alternatives did the software consider? Is the software’s decision correct? These are basic yet important questions with any software-driven approach – which is the bulk of today’s MS/MS data analysis. A lot of it comes down to trust, but trust has to be earned. With Mascot products, you can always drill down into the data and inspect the evidence for yourself.
Peptide identification is the foundation of bottom-up LC-MS/MS analysis. Mascot Server never relies on just one MS/MS fragment when identifying a peptide. The program starts by dividing the spectrum into 100Da bins and chooses the tallest peak in each bin. The peaks are matched to the theoretical fragment masses and scored probabilistically. The procedure is repeated in peak intensity order until the score has stopped improving. The algorithm and the scoring model try to explain the most prominent peaks across the whole MS/MS spectrum, and it’s robust against missing peaks and noise. On the other hand, there are cases where Mascot might not match and score all fragments if confident identification is possible with fewer peaks.

Figure 1: Screenshot of Spectrum Viewer embedded in Peptide View
from Variable Modifications in Mascot 2.7.
You can inspect the peak matching results for every peptide match in Peptide View. Mascot records the top 10 best matches for each spectrum, which can also be compared in Peptide View. It’s not practical to manually validate every rank 1 peptide match, but it’s a good idea to do spot checks. Fragmentation evidence may point to sources of systematic error or poor peak picking. Rank 2-10 matches may point out ambiguity with the sequence or variable mods, or issues with search parameters or choice of database.
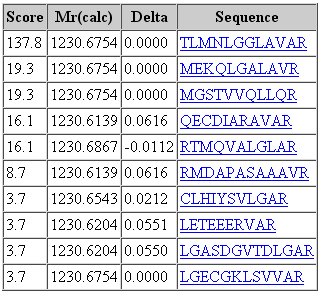
Figure 2: Screenshot of top 10 sequence matches to a spectrum
from High-mass accuracy: fragments.
Once these have been double checked and corrected, it’s worth looking at peptide match statistics at the search level. Probability-based scoring relies on certain assumptions that are not always true, sometimes phrased as all models are wrong, but some are useful. We recommend running a target-decoy search to calibrate the error rate, which adjusts for incorrect assumptions. If you submit the search as automatic decoy search, Mascot generates the decoy database on the fly.

Figure 3: Screenshot of the display and selection of PSM FDR
from Back to basics 5: Peptide-spectrum match statistics.
Search results for decoy peptide matches and decoy proteins are not hidden. In Protein Family Summary, expand the section for Sensitivity and FDR and click the link to view the decoy report. It can be instructive to compare the fragment mass evidence between target and decoy matches and to check whether false discovery rate (FDR) estimation has set the score threshold to a reasonable value.
Moving on to protein identification, the basic question is whether the protein hits presented by the software are supported by the peptide evidence. Mascot only uses confidently identified peptides for protein inference. These are the statistically significant peptide matches at the chosen PSM or sequence FDR. The hierarchical clustering algorithm groups proteins into families based on shared peptide matches. Proteins are separated by unique peptides, so each family member has at least one significant, unique peptide. Because of this, proteins that appear as family members are likely to be in the sample.

Figure 4: Screenshot of shared and unique alkaline phosphatase peptides
from the example error tolerant search.
By default, Mascot displays one-hit wonders, proteins identified from a single peptide match. You can always change the format control to require two, three or more peptides per protein. Protein FDR can be used for estimating the proportion of wholly false family members. Any peptide match assigned to a protein hit that is “discarded” in this way goes to the unassigned list.
A protein hit without unique peptide evidence is harder to assess. Protein Family Summary displays sameset and subset proteins by default. Two proteins are a sameset if they share exactly the same peptide matches. As there is no peptide-level evidence to say otherwise, either one or both of the sameset proteins could be in the sample. Mascot chooses one as the representative protein, but you can control this by setting preferred taxonomy. A protein is a subset or intersection protein if its peptide evidence is explained by two or more family members. Identifying a sameset or subset protein confidently requires an enzyme that produces a peptide unique to the protein, and the peptide must be detectable in MS/MS.
The key point is that you don’t need blind faith. You can always look at the peptide evidence for a protein identification to assess the quality of the search results and whether the software made the right call.
Finally, if you repeat the Mascot search, will you get the same results? Yes. As detailed in Is your database search reproducible?, Mascot is predictable, very stable and has few system dependencies. When (not if!) you need to find out exactly what was searched and how, all the required metadata to repeat the search is saved in the results file.
Keywords: FDR, interpretation, protein inference, reproducibility, scoring