Obsolete MS/MS summary reports
The Protein Family Summary report is the default summary report for MS/MS searches. This page describes obsolete MS/MS summary reports originally developed for smaller searches. These reports are still available, but they will be removed in a future Mascot release.
Protein inference
The obsolete reports use an old protein grouping algorithm described below.
First, we take the protein with the highest protein score, and call this hit number 1. We then take all other proteins that share the same set of peptide matches or a sub-set and include these in the same hit. In the report, they are listed as same-set and sub-set proteins. With these proteins removed from the list, we now take the remaining protein with the highest score and repeat the process until all the significant peptide matches are accounted for.
This sounds simple enough, and works well for small datasets, but larger search results create difficulties:
- What if two proteins have many strong matches in common but one has an additional weak match? Should we treat one as the outright winner, and relegate the other to the status of sub-set?
- What if we have intersections? That is, the protein is not a sub-set of any other one protein, but all the matches can be found in a set of proteins, each of which has additional matches.
- In many cases, the exact sequence of the protein that was analysed is not in the database. All the peptide sequences are present, but spread across several homologous proteins, which might be splice variants or represent different combinations of SNPs.
These issues are solved in the Protein Family Summary algorithm introduced in Mascot Server 2.3.
Peptide Summary (obsolete)
For MS/MS searches of less than 300 spectra, the Peptide Summary provides a clear picture of the peptide matches, grouped into protein hits using a simple parsimony algorithm. If there are 300 or more spectra, you should use the Protein Family Summary. This groups the proteins into families based on a novel hierarchical clustering algorithm. The Protein Family Summary report is ideally suited to very large and complex MS/MS searches, where it is not practical to display all the results on a single HTML page.
Sections of the report are described in the order in which they appear. Use this link to open an example report in a new browser window or tab.
Header
At the top of the report are a few lines to identify the search uniquely: search title, date, user name, etc. The database version is identified with either a release number or an ISO datestamp. The accessions and descriptions for the top scoring protein hits are listed.
If the search included the auto-decoy option, false discovery rate information is displayed at this location.
Score Distribution
Following the header, a histogram illustrates the protein score distribution. The 50 highest protein scores are divided into 16 bins according to their score, and the heights of the bars show the number of matches in each bin.
For a search of MS/MS data, protein scores are derived from ions scores as a non-probabilistic basis for ranking protein hits. The protein score histogram has little meaning for MS/MS results, but is retained for historical reasons. The green shaded area extends up to the average identity threshold for an individual peptide match.
Format Controls
These controls enable the report format to be modified. After making changes, press the "Format As" button to reload the report using the new settings.
- Report format Choose from the list of available formats
- Significance threshold The default significance threshold is p < 0.05. You can change this to any value in the range 0.99 to 1E-18.
- Maximum number of hits This value was initially chosen when the search was submitted. Enter a positive integer if you wish to re-specify the number of protein hits to report. Of course, the total number of hits actually found by the search may be less. Entering the word AUTO or a value of 0 will display all of the hits that have a protein score exceeding the average identity threshold score for an individual peptide match.
- Standard or MudPIT scoring MudPIT scoring is a more aggressive protein score that removes protein hits that have high protein scores purely because they have a large number of low-scoring peptide matches. It is the default protein score for a search in which the ratio of the number of queries to the number of entries in the database, (after any taxonomy filter), exceeds 0.001.
- Ions score cut-off Values greater than 0 and less than 1 act as an expect value threshold, and the scores for any peptide matches with higher expect values are suppressed. Values of 1 or more act as a score threshold, and any peptide matches with lower scores are suppressed. By setting this to (say) 20, you cut out all of the very low scoring, random peptide matches. This means that homologous proteins are more likely to collapse into a single hit.
- Show sub-sets By default, each hit in a Peptide Summary shows the set of proteins that match a particular set of peptides. Proteins that match a sub-set of those peptides are not shown. You can choose to show these additional protein hits, but be aware that this can make the report very much longer. To show all sub-set proteins, set Show sub-sets to 1. To show no sub-set proteins, set it to 0. Intermediate values set a threshold on the difference in protein score between the primary hit and the sub-set hit expressed as a fraction. For example, if the protein score of the primary hit was 400, and Show sub-sets was 0.75, sub-set hits with scores of 100 or more would be displayed.
- Show or Suppress pop-ups The JavaScript pop-up windows, that show the top 10 peptide matches for each query, are very useful, but they make the HTML report larger and slower to load in a web browser. If you have a report that never seems to load, or is very slow to scroll, try suppressing the pop-ups.
- Sort unassigned These are sorting options for the list of peptide matches that are not assigned to protein hits. Descending score makes it easy to see whether there are any good matches. If so, you will want to increase the number of protein hits or set it to AUTO so as to pull these matches into the main body of the report. Ascending query number is the same as ascending precursor Mr. Descending intensity allows you to find strong spectra that have failed to get a match. These could be candidates for de novo sequencing.
- Require bold red Requiring a protein hit to include at least one bold red peptide match is a good way to remove duplicate homologous proteins from a report.
The following controls are only displayed for certain types of search:
- UniGene index (Only displayed for a search of a nucleic acid database when UniGene index files have been configured) Choose the UniGene index to be used to cluster the protein hits into gene based families.
- Hide error tolerant matches (Only displayed for an automatic error tolerant search) Check to hide the additional, error tolerant matches.
- Show Percolator scores (Only displayed for searches that satisfy the requirements for using Percolator) Check to display scores and expect values adjusted by Percolator.
If the search used a quantitation method specifying Multiplex or Reporter protocol, there will be an additional block of quantitation related controls, as described here.
Repeating a search
A search can easily be repeated, so as to investigate the effect of changes in search parameters. Queries can selected in the result report, then loaded into a search form, where the search parameters can be modified.
Checkboxes for selecting queries for a repeat search are included in the body of the report, wherever the top rank match fir a particular query first appears. You can toggle individual checkboxes or use the Select All and Select None buttons to change the states of all checkboxes. Search Selected invokes the search form.
There is a second series of checkboxes, one for each protein hit, that have a dual purpose. If you wish to perform a manual error tolerant search, first check the Error Tolerant checkbox then select the proteins to be searched in the second pass search. Press Search Selected to invoke the search form.
If the Error Tolerant checkbox is not checked, the checkboxes select protein hits for an Archive Report
Protein Hit List
The body of the Peptide Summary report contains a tabular listing of the proteins, sorted by descending protein score. For each protein, the first line contains the accession string, (linked to the corresponding Protein View), the protein molecular mass, and the protein score. The number of queries matched to the protein completes the first line. The second line is the protein description taken from the Fasta entry. This is followed by a table summarising the matched peptide masses. The table columns contain:
- Checkboxes for selecting queries for a repeat search will appear in the first column of any row containing the first appearance of a top ranked match.
- Query number, hyperlinked to Peptide View.
- Experimental m/z value
- Experimental m/z transformed to a relative molecular mass
- Relative molecular mass calculated from the matched peptide sequence
- Difference (error) between the experimental and calculated masses
- Number of missed cleavage sites
- Ions score – If there are duplicate matches to the same peptide, then the lower scoring matches are shown in brackets.
- Expectation value for the peptide match. (The number of times we would expect to obtain an equal or higher score, purely by chance. The lower this value, the more significant the result).
- Rank of the ions match, (1 to 10, where 1 is the best match).
- A letter U if the peptide sequence is unique to the protein hit
- Sequence of the peptide in 1-letter code. The residues that bracket the peptide sequence in the protein are also shown, delimited by periods. If the peptide forms the protein terminus, then a dash is shown instead.
- Any variable modifications found in the peptide
If the search used a quantitation method specifying Multiplex or Reporter protocol, there will be an additional rows and columns of quantitation information, as described here.
An abbreviated listing follows for any proteins that contain the same set of peptide matches. It is also possible to display proteins containing a sub-set of peptide matches, but this is disabled by default. It can be enabled globally in the configuration file, mascot.dat, or enabled for a single report by using the checkbox in the format controls.
Yellow Pop-up
Clicking on the query number link opens the Peptide View for the match in a new browser window or tab. Resting the mouse cursor over the query number link causes a pop-up window to appear, displaying the complete list of peptide matches for that query. The pop-up window displays the query title (if any) followed by one or two significance thresholds, which are described in detail here. Below this, a table containing information on the highest scoring peptide matches for the query:
- Ions score
- Expect value
- Difference (error) between the experimental and calculated masses
- Hit number of the (first) protein containing the peptide match. A plus sign indicates that multiple proteins contain a match to this peptide
- Accession string of the (first) protein containing the peptide match.
- Sequence of the peptide in 1-letter code. If a variable modification has been used to obtain a match, the modified residue is underlined. If the residues that bracket the peptide sequence are the same in all the proteins that contain it, then these residues are also shown, delimited by periods. If the peptide forms the protein terminus, then a dash is shown instead.
Unassigned Peptide Matches
The unassigned list contains peptide matches that are not assigned to proteins in the body of the report. In some cases, there may be no match at all, and only the observed m/z value and the experimental Mr will be listed. In other cases, the top scoring peptide match will be listed.
So, unassigned doesn’t necessarily mean unmatched or not significant. Its the overflow, if you like. If you reformat the report, asking for more and more protein hits, all of the unassigned matches with non-zero scores would eventually get pulled into the body of the report.
When you load the peptide view for an unassigned query, it takes the first protein containing the matched peptide. This may or may not be the protein that would be selected as the primary hit if the peptide match was pulled into the body of the report.
Search Parameters
At the foot of the report, the search parameters are summarised. Descriptions of individual search parameters can be found here.
Select Summary (obsolete)
From Peptide Summary, you can use the format controls to switch to a Select Summary, which is similar to a Peptide Summary, but provides a more compact view of the results. The Select Summary splits the peptide matches assigned to protein hits into a separate report from the unassigned peptide matches.
Use this link to open an example report in a new browser window or tab.
The Select Summary was inspired by David Tabb’s DTASelect. It is very similar to the Peptide Summary, but more compact because multiple matches to the same peptide sequence are collapsed into a single line. Also, the list of peptide matches that are not assigned to any protein hit is split off into a separate report.
The differences between the Select Summary and the Peptide Summary are as follows:
- The Search Parameters are moved up into the header
- There is no Score Distribution histogram
- There are no checkboxes against protein hits or peptide matches. The choices for repeating a search are set by radio buttons for All queries, Unassigned, Below homology threshold, and Below identity threshold. This means that you cannot use a Select Summary for a manual error tolerant search or to create an Archive Report
- In the Protein Hit List, if multiple queries match to the same peptide (the same sequence, modifications and charge state) full details are displayed for the highest scoring match only. Any matches with lower scores are listed as additional query number links after the peptide sequence.
- Variable modifications are not listed after the peptide sequence, but are indicated by underlined residues.
- Unassigned Peptide Matches are split off into a separate report. Use the format controls to display this report.
Archive Report (obsolete)
If you are submitting MS/MS searches to an in-house Mascot server, you will also have the option to create an Archive Report. This is simply an edited version of the Peptide Summary report, that only includes the protein hits you have selected. If there are no peptide sequence matches at all from a search of MS/MS data, only molecular weight matches, then a Protein Summary report will be displayed. This indicates that the search has failed. Possibly the spectra are nothing but noise or possibly the search parameters are incorrect in some way.
Use this link to open an example report in a new browser window or tab.
An Archive Report is a Peptide Summary in which only selected protein hits appear. There is no unassigned list and no controls for changing the format or repeating the search. You might use this format to give a colleague a list of validated protein hits, or to remove hits that are contaminants or of no interest, etc.
Large Search Results and Legacy Reports
The Protein Family Summary is expressly designed for large search results. If, for some reason, you need to view results using the earlier, Select Summary report, this section contains some tips.
The format controls near the top of the report can help streamline the results from a large search by eliminating most of the "junk". If the report is too large to open in the first place, these options can also be specified by adding URL switches to the report URL.
- View the report on a client with plenty of free physical RAM. Do not try to view the report in a browser running on the Mascot server.
Select Summary: Ensure you are using the Select Summary. If you are using a third party client that has specified Peptide Summary,
Add this to the URL before opening the file: &REPTYPE=selectDon’t specify too many hits: Use AUTO to report only protein hits that contain significant peptide matches
Add this to the URL before opening the file: &REPORT=AUTOMudPIT Protein Scoring: By default, large searches will switch to using more aggressive protein scoring. This removes many of the junk protein hits, which have high protein scores but no high scoring peptide matches. Do not be tempted to switch back to standard scoring.
Add this to the URL before opening the file: &_server_mudpit_switch=0.000000001Require Bold Red: The Select Summary report does not detect intersections. Red and bold typefaces are used to highlight the most logical assignment of peptides to proteins. The first time a peptide match to a query appears in the report, it is shown in bold face. Whenever the top ranking peptide match appears, it is shown in red. Thus, a bold red match is the highest scoring match to a particular query listed under the highest scoring protein containing that match. This means that protein hits with many peptide matches that are both bold and red are the most likely assignments. Conversely, a protein that does not contain any bold red matches is an intersection of proteins listed higher in the report.
Requiring a protein hit to include at least one bold red peptide match is a good way to filter homologous proteins from a report. The down-side is that you may sometimes throw out the wrong protein! For example, imagine you are searching with a taxonomy of mammals but are mainly interested in yeti proteins. If the same strong peptide matches are found in a yeti protein and also in the human homolog, and one or more junk peptide matches prevent the two proteins collapsing into a single hit, but give the human protein a slightly higher score, that is the one that will feature in the report.
Add this to the URL before opening the file: &_requireboldred=1Ignore Ions Score Below: You can minimise the previous problem by judicious use of the Ions score cut-off field. By setting this to a value of 1 or more, you filter out all of the matches with lower scores. When set to a value between 0 and 1, it becomes an expect value cut-off, filtering out matches with higher expect values. Removing random matches means that homologous proteins are more likely to collapse into a single hit. (Note that this control is not displayed by default. The default control is a checkbox labelled Display non-significant matches which should be left unchecked. For more information, search the Installation & setup manual for DisplayNonSignificantMatches.)
Add this to the URL before opening the file: &_ignoreionsscorebelow=0.5Suppress the pop-ups: The JavaScript pop-up windows, that show the top 10 peptide matches for each query, are very useful, but they make the HTML report much larger and slower to load in a web browser. If you have a report that never seems to load, or is very slow to scroll, try using the radio buttons to suppress pop-ups.
Add this to the URL before opening the file: &_showpopups=FALSE
Manual Error Tolerant Search (obsolete)
It is possible to submit a ‘manual’ error tolerant search from Peptide Summary and Select Summary reports. This is a two-pass search similar to the automatic error tolerant search, except protein selection for the second pass is done manually. This was an earlier implementation, and is retained mainly for compatibility with existing workflows and third party software.
The manual error tolerant search should only be used in exceptional cases. One reason is that, because enzyme specificity is dropped entirely, and modifications can be combined with non-specificity, and the number of database entries tends to be fewer, the level of "junk" matches in the manual search will be higher than in the automatic search. Another reason is that, in the automatic search, the results from both passes are saved to the result file, which provides greater reporting flexibility. For example, you can choose to show or hide the additional, error tolerant matches. The combined report also reduces compatibility problems for applications that read Mascot result files.
Database entries are selected from the results report of a standard search. Check the Error tolerant checkbox, near the Search selected button, and choose one or more proteins to be included in the search. (On the public web site, a maximum of 3 proteins can be chosen). Clicking on the Search selected button loads a modified search form, from which you can change many of the search parameters. Cleavage agent defaults to None, though an enzyme can be chosen if desired.
During the error tolerant search:
- The complete list of modifications is tested, serially
- For a protein, the set of all possible amino acid substitutions is tested. For a nucleic acid sequence, all single base insertions, deletions, and substitutions are tested.
In the results report, protein scores derived are from all the matches listed, including error tolerant matches.
Quantitation report layout (Select Summary)
When the quantitation method is either Reporter or Multiplex, there are several additions to the Select Summary. Quantitation related information is presented on a yellow background, in four main areas: the header, the format controls, (described above), a quantitation summary for each protein hit, and ratios for individual peptides, (optional).
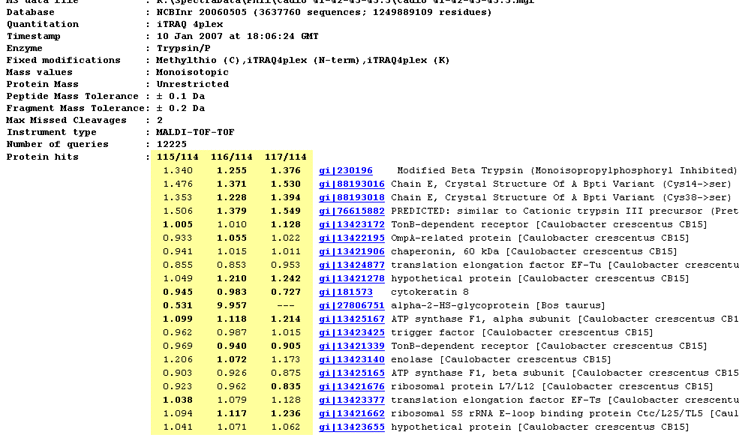
The report header includes a list of the protein hits tabulated in the body of the report. Where quantitation information is available, it is listed alongside each protein. The ratios to be reported and the column headings are specified as part of the quantitation method.
- If a ratio is shown in bold face, it is significantly different from 1 at a 95% confidence level. More details can be found on the Statistical procedures page. This may or may not be meaningful. In this screen shot, for example, the top hits are trypsin variants, so the quantitation information is simply irrelevant.
- If a ratio is missing, this will usually be because the number of valid peptide match ratios was less than the stipulated minimum.
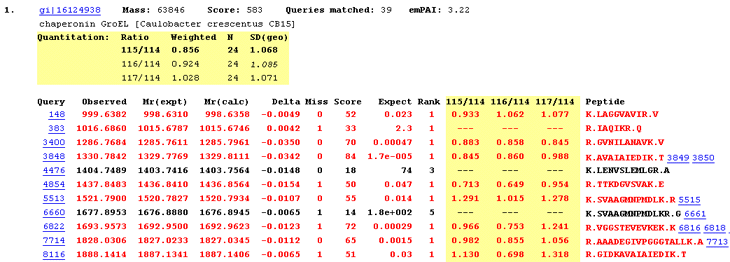
For each protein hit, a table shows the average ratio(s) for the protein, together with the number of peptide ratios that contributed, N, and the geometric standard deviation, SD(geo). If the number of valid peptide match ratios was less than the stipulated minimum, dashes will be displayed. If the peptide match ratios do not appear to be a sample from a normal distribution, the geometric standard deviation will be displayed in italics, and will never be bold, because it must be considered unreliable. As mentioned previously, bold face indicates a ratio that is significantly different from 1 at a 95% confidence level.
Since we are dealing with ratios, the average is the geometric mean and the standard deviation is the geometric standard deviation, which is a factor. In other words, the confidence interval is obtained by dividing and multiplying the average by the standard deviation, which is never less than 1.0. For example, if the average is 1.055 and SD(geo) is 1.028 then the 95% confidence interval is 1.026 to 1.084.
Warning: The standard deviation reported here measures the variance within the data submitted for a single Mascot search. This will often be tiny compared with the variance between technical replicates, (i.e. repeated analyses of this same sample). This, in turn, will invariably be tiny compared with the variance between biological replicates, (analyses of samples from different subjects or different treatment groups). It can be dangerous to read too much into small changes within a single search when the biological variance is huge. For a more detailed discussion, see Karp, N. A., et al., Impact of replicate types on proteomic expression analysis, Journal of Proteome Research 4 1867-1871 (2005).
Individual peptide match ratios will be displayed if the Report peptide ratios checkbox is checked. Dashes are displayed when a ratio cannot be determined. This may be because one or more of the relevant peaks were missing, giving a ratio which was zero, infinity, or indeterminate. Alternatively, the peptide match may have been rejected on quality grounds. For example, a disallowed charge state or modification. If a ratio is negative, which indicates some instrument or peak detection problem, (or an inappropriate correction), this is reported. However, a negative ratio is discarded when calculating the protein ratio.
The screen shot illustrates a Select Summary report, where detailed information is only displayed for the strongest match to each unique peptide sequence. The quantitation information also pertains to just the strongest match. It is not an average of all the matches in the row.
URL Switches
There are a number of switches to modify the format of the result reports. Many of these have a global default, set by a parameter in the Options section of mascot.dat. These defaults can be changed in an individual report using the format controls, or by appending the relevant switch to the report URL. Switches take the form label=value and the delimiter between switches is an ampersand (&). For example, if the report URL was:
http://local-server/mascot/cgi/master_results.pl?file=../data/20040121/F001847.dat
The type of report could be changed by appending "REPTYPE=protein":
http://local-server/mascot/cgi/master_results.pl?file=../data/20040121/F001847.dat&REPTYPE=protein
Labels and values are not case sensitive. Note that many labels begin with an underscore character. Values that are not literal strings are shown in italics.
master_results.pl
| URL | mascot.dat | master_results.pl | Value | Description |
|---|---|---|---|---|
| reptype | peptide | Peptide Summary | ||
| archive | Archive Report | |||
| concise | Concise Protein Summary | |||
| protein | Full Protein Summary | |||
| select | Select Summary (hits) | |||
| unassigned | Select Summary (unassigned) | |||
| report | auto | Report all significant hits | ||
| N | Report N hits | |||
| _showsubsets | ShowSubSets | 1 | For a Peptide Summary, set the value to 1 to report all hits that match a subset of peptides. Default is 0 for no sub-set hits. Intermediate values set a threshold on the difference in protein score between the primary hit and the sub-set hit expressed as a fraction. | |
| _requireboldred | RequireBoldRed | 1 | Set value to 1 to report Peptide Summary hits only if they contain at least one "bold red" peptide, (default 0). | |
| _showallfromerrortolerant | ShowAllFromErrorTolerant | 1 | Set value to 1 to report all matches from an error tolerant search, including the garbage, (default 0) | |
| _onlyerrortolerant | 1 | Set value to 1 to report only error tolerant matches from an automatic error tolerant search, (default 0) | ||
| _noerrortolerant | 1 | Set value to 1 to suppress error tolerant matches from an automatic error tolerant search, (default 0) | ||
| _show_decoy_report | 1 | Set value to 1 to display the report for an automatic decoy database search, (default 0) | ||
| _sigthreshold | SigThreshold | N | Probability to use for the significance threshold. Range is 0.99 to 1E-18, (default 0.05). | |
| _sortunassigned | SortUnassigned | scoredown | Sort unassigned matches by descending score, (default) | |
| queryup | Sort unassigned matches by ascending query number | |||
| intdown | Sort unassigned matches by descending intensity | |||
| _ignoreionsscorebelow | IgnoreIonsScoreBelow | N | Values greater than 0 and less than 1 act as an expect value threshold, and the scores for any peptide matches with higher expect values are set to 0, so that they disappear from the report. Values of 1 or more act as a score threshold, and any peptide matches with lower scores suppressed. A value of -1 means set the threshold to the value of _sigthreshold. Floating point number, (default 0.0). | |
| _showpopups | true | Show top 10 peptide matches for each query in JavaScript pop-up, (default) | ||
| false | Suppress JavaScript pop-ups. | |||
| _alwaysgettitle | 1 | Set to 1 to force reports to fetch Fasta titles from database when they are not included in the result file, (default 0 in master_results.pl, 1 in master_results_2.pl). | ||
| _server_mudpit_switch | MudpitSwitch | N | Protein score calculation switches to large search mode when the ratio between the number of queries and the number of database entries, (after any taxonomy filter), exceeds this value, (default 0.001). | |
| percolate | Percolator | 1 | Set value to 1 to re-rank results using Percolator, (default 0). | |
| percolate_rt | PercolatorUseRT | 1 | Set value to 1 to include retention time feature when using Percolator, (default 0). |
Quantitation
URL arguments relating to quantitation are described here