Mascot vs FragPipe: Uncovering endogenous proteolytic processing
Studying endogenous proteolytic processing, or N-terminomics, typically involves selective enrichment of protein N-termini. An alternative is to use the standard shotgun LC-MS/MS approach with the unenriched sample, which requires the database search to identify semi-specific peptides. Mascot Server 3.0 includes MS2PIP machine learning models for fragment intensity prediction, which can give a big boost to semi-specific identifications. Recent versions of FragPipe follow the same rescoring idea, with MSBoost providing the predictions. So which one performs better?
Terminomic murine polycistic kidney disease (PDK) model
We selected raw data from MassIVE project MSV000094661. This is the data behind the recent publication for TermineR, which is a pipeline for analyzing endogenous proteolytic processing. Specifically, we selected raw data from the second experiment in the publication, where the authors process TMTpro-labelled samples from a terminomic murine polycystic kidney disease (PDK) model. There were 6 kidney tissue samples presenting enlarged cystic kidneys and five wild type samples.
The TMTpro labeling was performed at the protein level, so we need to treat N-terminal labelled peptides as variable. By labelling the sample prior to digestion, all the free protein N-terminal sites will be labelled with TMTpro, and their identification will uncover any unsuspected proteolytic processing.
As the N-terminal peptides won’t necessarily conform to tryptic cleavage rules, a semi-specific enzyme needs to be used. Also, trypsin cannot cleave at the TMTpro-labelled lysines. I created a semi-ArgC enzyme to model these constraints for the database searching. N-terminal acetylation was also selected as a variable modification at the peptide level (N-term) rather than protein level (Protein N-term) in order to identify N-terminals with unsuspected proteolytic processing and acetylation.
We used three databases for searching: the Uniprot UP589_M_musculus proteome, the contaminants database (containing trypsin, human keratin and other common contaminants) and a custom database with the Biognosys_iRT peptides. The rest of the search parameters were the same as the publication.
The final search parameters looked like this:
Enzyme : SemiArgC Fixed modifications : Carbamidomethyl (C), TMTpro (K) Variable modifications : Acetyl (N-term), TMTpro (N-term) Mass values : Monoisotopic Protein mass : Unrestricted Peptide mass tolerance : ± 20 ppm Fragment mass tolerance : ± 20 ppm Max missed cleavages : 1 Instrument type : ESI-TRAP
While working on the data, we noticed that there were a lot of TMTpro complementary reporter ion clusters in the spectra. To mitigate this, we used the script TMT_complementary_ions.pl to remove any complementary reporter ions, which gives a slight boost to the peptide scoring and identification for those spectra. Another option is to convert the complementary ions into reporter ions, but this is not possible due to the choice of TMTpro labels used in the experiment – some of the channels overlap.
Results
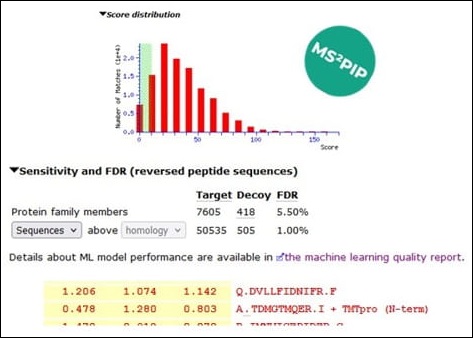
We ran the search using Mascot Server 3.0, enabled refining with machine learning and selected the MS2PIP TMT model. The other MS2PIP models are not suitable for TMTpro-labelled peptides, as the TMTpro modification significantly alters the fragmentation pattern.
(Mascot also ships with a DeepLC TMT model for retention time predictions (full_hc_hela_hf_psms_aligned), but we found it did not improve the results sufficiently to justify the extra computation, and the other DeepLC models were not suitable with these data either.)
| FragPipe with MSBoost |
Mascot with MS2PIP TMT model |
|
|---|---|---|
| Protein hits | 7560 | 7605 |
| Unique sequences | 44470 | 50535 |
| TMTpro N-term | 3062 | 3219 |
| Acetyl N-term | 617 | 883 |
The FragPipe counts come from the FragPipe protein and peptide .tsv reports, as downloaded from the MassIVE repository. The spreadsheets appear to be thresholded to 1% FDR.
For the Mascot Server results, we took the protein and sequence counts from the FDR statistics section of the report. The results were thresholded to 1% sequence FDR, and every protein hit reported by Mascot is supported by unique peptide evidence. The results were then exported to CSV.
We opened both spreadsheets in Excel. After some filtering and a pivot in Excel, we can count the Acetyl N-term and TMTpro N-terminal matches. We did not include protein N-terminal peptides (preceding residue ‘-’), but did include all the peptides with Methionine as the preceding N-terminal residue. For some of these, the Methionine will be at protein position 1 and has been removed during post-translational processing, so it is actually the natural protein N-terminal. The majority of these peptides are the semi-specific peptides from ragged protein N-terminals prior to digestion.
In conclusion, Mascot Server 3.0 using MS2Rescore and the MS2PIP TMT model performed eminently and is a good choice for the analysis for N-terminomics.
Finally, how long does the computational processing take? The raw files converted into nearly 600k queries (MS/MS spectra), and the semi-specific database search with Mascot Server took 303 seconds on my 16 core server (AMD Ryzen 9 9950X). Machine learning took another few minutes at the end. Although semi-specific enzyme makes the search space a lot bigger, the mouse proteome has just 63,149 protein sequences. This is tiny relative to what Mascot is designed to do.
Keywords: machine learning, MS2PIP, Percolator, TMT